催化剂及其制备方法、异氰酸酯的聚合方法与流程
1.本发明涉及多异氰酸酯聚合领域,具体涉及一种催化剂及其制备方法、异氰酸酯的聚合方法。
背景技术:
2.二异氰酸酯类聚合物在各种聚氨酯类涂料和胶粘剂中有着其独特的优势而被广泛地使用。但单体易挥发且有毒性,因此多将其制备为多聚体使用。由于脂肪族二异氰酸酯的聚异氰脲酸酯具有很好的热稳定性、耐候性以及耐介质性,其自聚反应被广泛的研究。制备过程中的难点在于催化剂的选择,目前使用的催化剂类型繁多,包括:季铵碱、季铵盐、金属盐、氢化物、氧化物等等。因此,如何利用绿色温和的反应条件来制备一种新型的催化剂,并保证其催化的异氰脲酸酯聚合反应有较高的单体转化率,且产物中三聚体含量较高是一个亟待解决的问题。
技术实现要素:
3.本发明所要解决的技术问题是:在催化剂中引入环状哌嗪骨架,从而使反应的单体转化率高、产物中三聚体占主要成分。
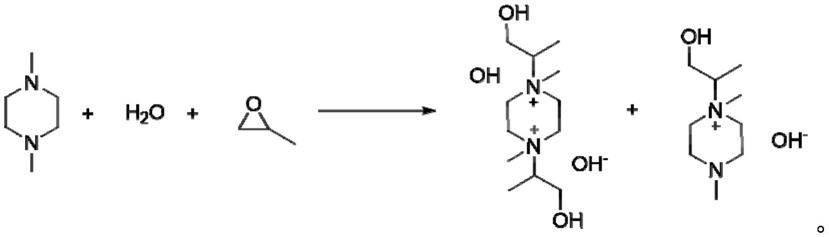
4.本发明解决其技术问题所采用的技术方案是:第一方面,所述催化剂的结构通式如下所示:
[0005][0006]
进一步的,所述催化剂的结构通式中r1为具有1
‑
20个碳原子的烷基。
[0007]
进一步的,所述催化剂的结构通式中r2为具有1
‑
20个碳原子的羟烷基。
[0008]
进一步的,所述催化剂的原料包括哌嗪的脂肪族取代衍生物、水、环氧烷烃;
[0009]
进一步的,所述哌嗪的脂肪族取代衍生物包括1,4
‑
二甲基哌嗪;所述环氧烷烃包括环氧丙烷。
[0010]
第二方面,所述催化剂的制备方法包括:在ar保护下,将一定量的环氧丙烷滴加到1,4
‑
二甲基哌嗪的水溶液中,其中水的用量为1,4
‑
二甲基哌嗪摩尔量的1.5
‑
4倍,环氧丙烷的用量为1,4
‑
二甲基哌嗪摩尔量的1.0
‑
2.5倍;然后在室温下搅拌,反应4
‑
9h;反应结束后取下层溶液;旋蒸得到哌嗪类季铵碱。
[0011]
主反应方程式如下所示:
[0012][0013]
第三方面,所述异氰酸酯的聚合方法包括:在惰性气体的保护下,以异氰酸酯为底物并在催化剂的催化作用下进行聚合反应,其中,反应温度为10
‑
100℃(优选30
‑
80℃),催化剂的用量以底物的质量为基准,其中催化剂的用量为10
‑
800ppm(优选200
‑
800ppm);本发明中使用的催化剂可在无溶剂情况下或溶解于催化剂溶剂中以溶液形式使用,当催化剂以溶液形式使用时,催化剂溶液的量占反应溶液量的5
‑
50wt%(优选10
‑
30wt%);随着反应的进行,当反应液中
‑
nco含量占反应液量的5
‑
80wt%(优选10
‑
70wt%或20
‑
50wt%)时,对反应采用高温处理或加入催化剂毒物使反应终止,其中,催化剂毒物的加入量为催化剂的两倍摩尔量或者两倍微过量。
[0014]
进一步的,适于本发明的催化剂溶剂包括含1~20个碳原子的直链或支链一元醇和二元醇,并且在分子整体中包含一个或多个羟基基团;可选的催化剂溶剂包括甲醇、乙醇、1
‑
丙醇或2
‑
丙醇、正丁醇、异丁醇、仲丁醇、叔丁醇、正辛醇、异辛醇、庚醇、2
‑
乙基
‑
1,3
‑
己二醇、1,3
‑
丁二醇或1,4
‑
丁二醇、1
‑
甲氧基
‑2‑
丙醇,优选采用甲醇、正丁醇、己醇、庚醇和异辛醇。
[0015]
进一步的,适于本发明的底物包括所有脂族异氰酸酯,其中,底物可以是单一的脂族异氰酸酯或者是多个脂族异氰酸酯的混合物;以下列出的所有异氰酸酯的结构或构型异构体均为可选实例:双(异氰酸根合烷基)醚、丙烷二异氰酸酯、丁烷二异氰酸酯、戊烷二异氰酸酯(五亚甲基二异氰酸酯,pdi)、己烷二异氰酸酯(六亚甲基二异氰酸酯,hdi)、庚烷二异氰酸酯、辛烷二异氰酸酯(例如,八亚甲基二异氰酸酯)、壬烷二异氰酸酯(例如,三甲基
‑
hdi、tmdi,通常作为2,4,4
‑
异构体和2,2,4
‑
异构体的混合物存在)、三异氰酸酯(例如,4
‑
异氰酸根合甲基
‑
1,8
‑
辛烷二异氰酸酯)、癸烷二异氰酸酯(例如,十亚甲基二异氰酸酯)和三异氰酸酯、十一烷二异氰酸酯和三异氰酸酯、十二烷二异氰酸酯(例如,十二亚甲基二异氰酸酯)和三异氰酸酯、十四烷二异氰酸酯(例如,十四亚甲基二异氰酸酯)、1,4
‑
环己烷二异氰酸酯(chdi)、3
‑
异氰酸根合甲基
‑
3,5,5
‑
三甲基环己基异氰酸酯(异佛尔酮二异氰酸酯,ipdi)、4,4
’‑
二环己基甲烷二异氰酸酯(h12mdi),优选采用五亚甲基二异氰酸酯(pdi)。
[0016]
进一步的,适于本发明的催化剂毒物包括酸或酸的衍生物;可选的催化剂毒物包括磷酸、苯甲酰氯、苯甲磺酸酯、磷酸酯、亚磷酸酯、甲磺酸和对甲苯磺酸等中的一种或多种,优选采用磷酸二丁酯。
[0017]
本发明的有益效果是:
[0018]
本发明在催化剂中引入环状哌嗪骨架,促进了小分子之间的反应,所述小分子反应包括异氰酸酯类底物的二聚反应和三聚反应,以及异氰酸酯类底物单体与体系中少量醇的反应,同时抑制大分子与小分子或大分子之间的反应,包括所生成聚合物与体系中少量醇的反应以及异氰酸酯类底物的多聚反应,从而达到单体转化率高、产物中三聚体占主要
成分的目的。
附图说明
[0019]
下面结合附图和实施例对本发明作进一步说明。
[0020]
图1是gpc测试结果曲线图。
[0021]
图中:a
‑
市售pdi三聚体,b
‑
本发明制得的pdi三聚体。
具体实施方式
[0022]
通过以下实施例将本发明所提供的方法予以进一步的说明,但本发明并不因此而受任何限制。本发明中涉及的所有百分数,除特殊说明外,均为质量百分数。
[0023]
实施例1
[0024]
调整1,4
‑
二甲基哌嗪、水、环氧丙烷摩尔比为1:1.5:1。在ar保护下,将环氧丙烷(3.5ml)在0.5h内滴加到的1,4
‑
二甲基哌嗪(6.8ml)和水(1.4ml)中。然后在室温下连续搅拌4h,得到反应产物1#。将合成得到的反应产物采用核磁进行定性分析。核磁结果如下所示:
[0025]
单取代季铵碱1h nmr(400m,tms):δ1.18(m,3h),1.89(s,3h),2.22
‑
2.50(m,10h),2.31(s,3h),3.15
‑
3.61(m,1h),5.6(s,1h)。
[0026]
二取代季铵碱1h nmr(400m,tms):δ1.06(m,6h),2.25
‑
2.31(m,8h),2.69(s,6h),3.36
‑
4.30(m,6h),5.6(s,2h)。
[0027]
进而对反应产物采用反相液相色谱进行定量分析,结果表明:单取代季铵碱质量含量为35.1%,二取代季铵碱质量含量为31.1%,副产物质量含量为33.8%。
[0028]
实施例2
[0029]
调整1,4
‑
二甲基哌嗪、水、环氧丙烷摩尔比为1:2:1.4。在ar保护下,将环氧丙烷(4.9ml)在0.5h内滴加到的1,4
‑
二甲基哌嗪(6.8ml)和水(1.9ml)中。然后在室温下连续搅拌4h,得到反应产物2#。将合成得到的反应产物采用核磁进行定性分析。核磁结果如下所示:
[0030]
单取代季铵碱1h nmr(400m,tms):δ1.18(m,3h),1.89(s,3h),2.22
‑
2.50(m,10h),2.31(s,3h),3.15
‑
3.61(m,1h),5.6(s,1h)。
[0031]
二取代季铵碱1h nmr(400m,tms):δ1.06(m,6h),2.25
‑
2.31(m,8h),2.69(s,6h),3.36
‑
4.30(m,6h),5.6(s,2h)。
[0032]
进而对反应产物采用反相液相色谱进行定量分析,结果表明:单取代季铵碱质量含量为19.1%,二取代季铵碱质量含量为78.5%,副产物质量含量为2.4%。
[0033]
实施例3
[0034]
调整1,4
‑
二甲基哌嗪、水、环氧丙烷摩尔比为1:2:1.4。在ar保护下,将环氧丙烷(4.9ml)在0.5h内滴加到的1,4
‑
二甲基哌嗪(6.8ml)和水(1.9ml)中。然后在室温下连续搅拌6h,得到反应产物3#。将合成得到的反应产物采用核磁进行定性分析。核磁结果如下所示:
[0035]
单取代季铵碱1h nmr(400m,tms):δ1.18(m,3h),1.89(s,3h),2.22
‑
2.50(m,10h),2.31(s,3h),3.15
‑
3.61(m,1h),5.6(s,1h)。
[0036]
二取代季铵碱1h nmr(400m,tms):δ1.06(m,6h),2.25
‑
2.31(m,8h),2.69(s,6h),3.36
‑
4.30(m,6h),5.6(s,2h)。
[0037]
进而对反应产物采用反相液相色谱进行定量分析,结果表明:单取代季铵碱质量含量为7.7%,二取代季铵碱质量含量为91.6%,副产物质量含量为0.7%。
[0038]
实施例4
[0039]
调整1,4
‑
二甲基哌嗪、水、环氧丙烷摩尔比为1:2:1.4。在ar保护下,将环氧丙烷(4.9ml)在0.5h内滴加到的1,4
‑
二甲基哌嗪(6.8ml)和水(1.9ml)中。然后在室温下连续搅拌9h,得到反应产物4#。将合成得到的反应产物采用核磁进行定性分析。核磁结果如下所示:
[0040]
单取代季铵碱1h nmr(400m,tms):δ1.18(m,3h),1.89(s,3h),2.22
‑
2.50(m,10h),2.31(s,3h),3.15
‑
3.61(m,1h),5.6(s,1h)。
[0041]
二取代季铵碱1h nmr(400m,tms):δ1.06(m,6h),2.25
‑
2.31(m,8h),2.69(s,6h),3.36
‑
4.30(m,6h),5.6(s,2h)。
[0042]
进而对反应产物采用反相液相色谱进行定量分析,结果表明:单取代季铵碱质量含量为9.2%,二取代季铵碱质量含量为44.7%,副产物质量含量为46.1%。
[0043]
实施例5
[0044]
调整1,4
‑
二甲基哌嗪、水、环氧丙烷摩尔比为1:2:2。在ar保护下,将环氧丙烷(7.0ml)在0.5h内滴加到的1,4
‑
二甲基哌嗪(6.8ml)和水(1.8ml)中。然后在室温下连续搅拌4h,得到反应产物5#。将合成得到的反应产物采用核磁进行定性分析。核磁结果如下所示:
[0045]
单取代季铵碱1h nmr(400m,tms):δ1.18(m,3h),1.89(s,3h),2.22
‑
2.50(m,10h),2.31(s,3h),3.15
‑
3.61(m,1h),5.6(s,1h)。
[0046]
二取代季铵碱1h nmr(400m,tms):δ1.06(m,6h),2.25
‑
2.31(m,8h),2.69(s,6h),3.36
‑
4.30(m,6h),5.6(s,2h)。
[0047]
进而对反应产物采用反相液相色谱进行定量分析,结果表明:单取代季铵碱质量含量为19.6%,二取代季铵碱质量含量为78.3%,副产物质量含量为2.1%。
[0048]
实施例6
[0049]
调整1,4
‑
二甲基哌嗪、水、环氧丙烷摩尔比为1:2:2.5。在ar保护下,将环氧丙烷(8.8ml)在0.5h内滴加到的1,4
‑
二甲基哌嗪(6.8ml)和水(0.1mol,1.8ml)中。然后在室温下连续搅拌4h,得到反应产物6#。将合成得到的反应产物采用核磁进行定性分析。核磁结果如下所示:
[0050]
单取代季铵碱1h nmr(400m,tms):δ1.18(m,3h),1.89(s,3h),2.22
‑
2.50(m,10h),2.31(s,3h),3.15
‑
3.61(m,1h),5.6(s,1h)。
[0051]
二取代季铵碱1h nmr(400m,tms):δ1.06(m,6h),2.25
‑
2.31(m,8h),2.69(s,6h),3.36
‑
4.30(m,6h),5.6(s,2h)。
[0052]
进而对反应产物采用反相液相色谱进行定量分析,结果表明:单取代季铵碱质量含量为3.7%,二取代季铵碱质量含量为93.4%,副产物质量含量为2.9%。
[0053]
实施例7
[0054]
调整1,4
‑
二乙基哌嗪、水、环氧丙烷摩尔比为1:2.5:2.5。在ar保护下,将环氧丙烷
(8.8ml)在0.5h内滴加到的1,4
‑
二乙基哌嗪(6.8ml)和水(2.3ml)中。然后在室温下连续搅拌4h,得到反应产物7#。将合成得到的反应产物采用核磁进行定性分析。核磁结果如下所示:
[0055]
单取代季铵碱1h nmr(400m,tms):δ1.18(m,3h),1.89(s,3h),2.22
‑
2.50(m,10h),2.31(s,3h),3.15
‑
3.61(m,1h),5.6(s,1h)。
[0056]
二取代季铵碱1h nmr(400m,tms):δ1.06(m,6h),2.25
‑
2.31(m,8h),2.69(s,6h),3.36
‑
4.30(m,6h),5.6(s,2h)。
[0057]
进而对反应产物采用反相液相色谱进行定量分析,结果表明:单取代季铵碱质量含量为9.1%,二取代季铵碱质量含量为88.6%,副产物质量含量为2.3%。
[0058]
实施例8
[0059]
调整1,4
‑
二甲基哌嗪、水、环氧丙烷摩尔比为1:4:2.5。在ar保护下,将环氧丙烷(8.8ml)在0.5h内滴加到的1,4
‑
二甲基哌嗪(6.8ml)和水(3.6ml)中。然后在室温下连续搅拌4h,得到反应产物8#。将合成得到的反应产物采用核磁进行定性分析。核磁结果如下所示:
[0060]
二取代季铵碱1h nmr(400m,tms):δ1.06(m,6h),2.25
‑
2.31(m,8h),2.69(s,6h),3.36
‑
4.30(m,6h),5.6(s,2h)。
[0061]
进而对反应产物采用反相液相色谱进行定量分析,结果表明:单取代季铵碱质量含量为0%,二取代季铵碱质量含量为98.3%,副产物质量含量为1.7%。
[0062]
分别利用上述实施例1
‑
8得到的反应产物作为催化剂以pdi为底物制备pdi的三聚体,如以下实施例9
‑
12所示:
[0063]
实施例9
[0064]
称取pdi(20g)单体加入反应瓶中,在ar保护下升温至90℃,预热一个小时后,控制温度稳定在80℃,滴加混合均匀的1#催化剂溶液600ppm,(采用甲醇进行稀释,稀释度为10%),滴加速度为1滴/15s,控制反应温度浮动范围不超过
±
1℃。当反应液中
‑
nco值达到40wt%左右时,立即加入初始催化剂两倍摩尔量的磷酸二丁酯终止反应。
[0065]
实施例10
[0066]
称取pdi(20g)单体加入反应瓶中,在ar保护下升温至90℃,预热一个小时后,控制温度稳定在80℃,滴加混合均匀的2#催化剂溶液600ppm,(采用甲醇进行稀释,稀释度为10%),滴加速度为1滴/15s,控制反应温度浮动范围不超过
±
1℃。当反应液中
‑
nco值达到40wt%左右时,立即加入初始催化剂两倍摩尔量的磷酸二丁酯终止反应。
[0067]
实施例11
[0068]
称取pdi(20g)单体加入反应瓶中,在ar保护下升温至90℃,预热一个小时后,控制温度稳定在80℃,滴加混合均匀的3#催化剂溶液600ppm,(采用甲醇进行稀释,稀释度为10%),滴加速度为1滴/15s,控制反应温度浮动范围不超过
±
1℃。当反应液中
‑
nco值达到40wt%左右时,立即加入初始催化剂两倍摩尔量的磷酸二丁酯终止反应
[0069]
实施例12
[0070]
称取pdi(20g)单体加入反应瓶中,在ar保护下升温至90℃,预热一个小时后,控制温度稳定在80℃,滴加混合均匀的4#催化剂溶液600ppm,(采用甲醇进行稀释,稀释度为10%),滴加速度为1滴/15s,控制反应温度浮动范围不超过
±
1℃。当反应液中
‑
nco值达到
40wt%左右时,立即加入初始催化剂两倍摩尔量的磷酸二丁酯终止反应。
[0071]
实施例13
[0072]
称取pdi(20g)单体加入反应瓶中,在ar保护下升温至90℃,预热一个小时后,控制温度稳定在80℃,滴加混合均匀的5#催化剂溶液600ppm,(采用甲醇进行稀释,稀释度为10%),滴加速度为1滴/15s,控制反应温度浮动范围不超过
±
1℃。当反应液中
‑
nco值达到40wt%左右时,立即加入初始催化剂两倍摩尔量的磷酸二丁酯终止反应。
[0073]
实施例14
[0074]
称取pdi(20g)单体加入反应瓶中,在ar保护下升温至90℃,预热一个小时后,控制温度稳定在80℃,滴加混合均匀的6#催化剂溶液600ppm,(采用甲醇进行稀释,稀释度为10%),滴加速度为1滴/15s,控制反应温度浮动范围不超过
±
1℃。当反应液中
‑
nco值达到40wt%左右时,立即加入初始催化剂两倍摩尔量的磷酸二丁酯终止反应。
[0075]
实施例15
[0076]
称取pdi(20g)单体加入反应瓶中,在ar保护下升温至90℃,预热一个小时后,控制温度稳定在80℃,滴加混合均匀的7#催化剂溶液600ppm,(采用甲醇进行稀释,稀释度为10%),滴加速度为1滴/15s,控制反应温度浮动范围不超过
±
1℃。当反应液中
‑
nco值达到40wt%左右时,立即加入初始催化剂两倍摩尔量的磷酸二丁酯终止反应。
[0077]
实施例16
[0078]
称取pdi(20g)单体加入反应瓶中,在ar保护下升温至90℃,预热一个小时后,控制温度稳定在80℃,滴加混合均匀的8#催化剂溶液600ppm,(采用甲醇进行稀释,稀释度为10%),滴加速度为1滴/15s,控制反应温度浮动范围不超过
±
1℃。当反应液中
‑
nco值达到40wt%左右时,立即加入初始催化剂两倍摩尔量的磷酸二丁酯终止反应。
[0079]
对比例
[0080]
称取pdi(20g)单体加入反应瓶中,在ar保护下升温至90℃,预热一个小时后,控制温度稳定在80℃,滴加混合均匀的2
‑
羟丙基三甲基辛酸铵盐(9#)溶液600ppm,(采用甲醇进行稀释,稀释度为10%),滴加速度为1滴/15s,控制反应温度浮动范围不超过
±
1℃。当反应液中
‑
nco值达到40wt%左右时,立即加入初始催化剂两倍摩尔量的磷酸二丁酯终止反应。
[0081]
本发明按照盐酸
‑
二正丁胺滴定法测定
‑
nco含量。本发明按照gb/t3143
‑
1982的方法测试产品色号。本发明使用凝胶渗透色谱(agilent
‑
1260,色谱柱plgel mixed
‑
c,流动相四氢呋喃)测定聚合物含量。本发明合成催化剂过程使用的试剂均购买自aladdin,如未特别说明,均为分析纯。实施例1
‑
8中的1,4
‑
二甲基哌嗪、水、环氧丙烷的用量如表1所示;对实施例9
‑
16和对比例所得聚合反应产物进行nco含量以及色度测试,结果见表2。
[0082]
表1.实施例1
‑
8实验数据表
[0083][0084]
表2.实施例9
‑
16以及对比例的实验数据表
[0085][0086]
由表1、表2,实施例2
‑
4中制备催化剂所用1,4
‑
二甲基哌嗪与水与环氧丙烷的比例均为1:2:1.4,反应时间分别为4、6、9依次升高,将实施例2
‑
4所制备的催化剂2#、3#以及4#分别用于实施例9
‑
11参与pdi聚合反应,当反应溶液中
‑
nco的比重达到40wt%左右时,所生成三聚体的含量较高,向反应体系中加入催化剂毒物,以将催化剂淬灭,终止聚合反应。实施例9
‑
11中完成聚合反应的时间分别为1h、1.7h、1h,产物pdi三聚体的色号分别为85、40、90,以上数据表明当催化剂在制备时所用底物比例相同的条件下,当制备时间为4h和9h时,所得催化剂的活性较高,但是所得pdi三聚体的色号较高,即制得的pdi三聚体发黄,产品的品质较低,制备时间为6h的催化剂的活性虽然偏低,但是其制备的pdi三聚体的品质远高于制备时间为4h和9h的催化剂。
[0087]
由表1、表2,实施例2、以及实施例5
‑
6中制备催化剂所用的时间均为4h并且其反应液中1,4
‑
二甲基哌嗪与水的比重也相同,反应液环氧丙烷的比重依次升高,当制得的催化剂2#、5#以及6#应用于pdi聚合反应,实施例9、实施例12以及实施例13中完成聚合反应的时间分别为1h、1.5h、4.2h,随着催化剂制备的反应体系中环氧丙烷的比例的升高,其制得的催化剂的活性下降。实施例7中相较于实施例6将反应液中水的比例升高制得7#,将7#应用于实施例14,数据表明实施例14中聚合反应完成的时间为2h远低于实施例13的4.2h,进一步表明在制备催化剂时反应体系中水的比重升高会提高催化剂的活性。
[0088]
经过对比,在实施例3条件下反应生成的催化剂3#应用于实施例10催化反应效果最好;实施例10中完成聚合反应的时间为1.7h,色号为40;对比例中聚合反应完成的时间为4h,色号为40;与对比例相比实施例10在保证聚合产物品质的前提下,其聚合反应完成的时间远低于对比例所用的时间。此外,还对在本实施例10条件下制得的pdi三聚体与市售的pdi三聚体做了凝胶渗透色谱实验,如图1所示,图中两条曲线的最高峰为pdi三聚体的峰,其余峰为多聚体的峰,并且峰的面积对应物质的含量;从gpc结果可以看出,本发明中三聚体含量(69.1%)与市售pdi三聚体含量(75.9%)相近。综上所述,本发明在催化剂中引入环状哌嗪骨架,促进了小分子之间的反应,如异氰酸酯类底物的二聚反应和三聚反应,以及异氰酸酯类底物单体与体系中少量醇的反应,同时抑制大分子与小分子或大分子之间的反应,如所生成聚合物与体系中少量醇的反应以及异氰酸酯类底物的多聚反应,从而达到单体转化率高、三聚体占主要成分的目的。
[0089]
以上述依据本发明的理想实施例为启示,通过上述的说明内容,相关的工作人员完全可以在不偏离本发明的范围内,进行多样的变更以及修改。本项发明的技术范围并不局限于说明书上的内容,必须要根据权利要求范围来确定其技术性范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1