检测替格瑞洛中(1R,2S)-2-(3,4-二氟苯基)环丙胺的方法与流程
本发明属于药物分析领域,具体涉及一种替格瑞洛基因毒性杂质(1r,2s)-2-(3,4-二氟苯基)环丙胺的含量检测方法。
背景技术:
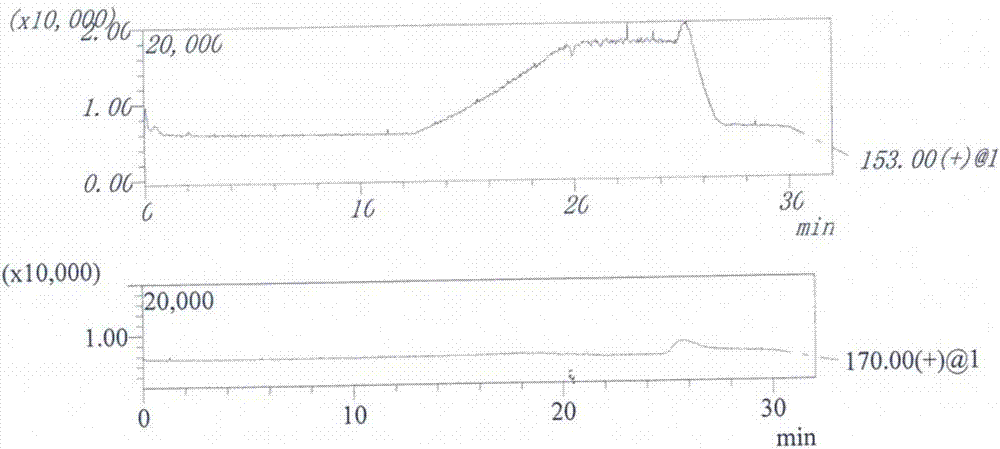
:替格瑞洛,通用名称:替格瑞洛;英文名称:ticagrelor;汉语拼音:tigeruiluo;化学名称为:(1s,2s,3r,5s)-3-[7-[[(1r,2s)-2-(3,4-二氟苯基)环丙基]氨基]-5-(丙硫基)-3h-1,2,3-三唑并[4,5-d]嘧啶-3-基]-5-(2-羟基乙氧基)-1,2-环戊二醇。替格瑞洛是由英国阿斯利康公司研制开发的一种新的治疗急性冠状动脉综合征的药物,2011年美国fda批准上市。其是选择性p2y12受体抑制剂,通过抑制新血凝块的形成,帮助患者降低动脉粥样化血栓形成事件的发生。急性冠状动脉综合征(acs)已成为威胁人类健康的疾病。血小板受体拮抗剂作为抗血小板聚集药,在acs的治疗过程中发挥了重要的作用。因此,替格瑞洛与氯吡格雷虽同为血小板抑制剂,但替格瑞洛临床试验表明安全性和有效性都更优,具有替代氯吡格雷的潜力,所以市场潜力巨大,应用前景广阔。目前,替格瑞洛主要通过以下路线合成:式中,r1为f、br、i、oh或oa,其中a为羟基活化基。其中,式ⅴ所示的化合物为(1r,2s)-2-(3,4-二氟苯基)环丙胺(r)-扁桃酸盐,是合成替格瑞洛的起始原料。由于受到工艺技术和控制水平的限制,生产得到的替格瑞洛产品中,很容易存在有未反应的作为杂质存在的(1r,2s)-2-(3,4-二氟苯基)环丙胺。(1r,2s)-2-(3,4-二氟苯基)环丙胺具有致癌性警示结构,根据ichq3a(r2)、m7以及文献1(文献1:romualdobenigni,ceciliabossa,olgatcheremenskaia.developmentofstructuralalertsfortheinvivomicronucleusassayinrodents[j].jrcscientificandtechnicalreports,24.),文献2(文献2:替格瑞洛片(jxhl1000319、320)国际多中心临床试验申请药学审评报告.)中明确提到该物质为基因毒性杂质,需要将替格瑞洛中的(1r,2s)-2-(3,4-二氟苯基)环丙胺的含量控制在7ppm以内,保证每日总摄入量不超过1.5μg;因而,十分有必要对替格瑞洛中(1r,2s)-2-(3,4-二氟苯基)环丙胺含量进行监控。cn105424822a报道了采用高效液相色谱法,以十八烷基硅烷键合硅胶为填充剂,流动相a为0.05mol/l磷酸盐缓冲液,b为乙腈,梯度洗脱的方法,为解决分离检测替格瑞洛中(1r,2s)-2-(3,4-二氟苯基)环丙胺提供一种思路,但该方法采用较低的检测波长(210nm)、梯度洗脱、较高的供试品溶液浓度(10mg/ml)、采用保留时间定性等必将导致专属性不强,结果重现性差等问题。本发明提供了一种新的液相色谱-质谱联用(lc-ms)的检测方法,用于检测替格瑞洛中的基因毒性杂质(1r,2s)-2-(3,4-二氟苯基)环丙胺,该方法专属性强,灵敏度高,为检测该基因毒性杂质含量提供了一种行之有效的方法,进一步保证了替格瑞洛的产品质量和患者的用药安全。技术实现要素:本发明的目的在于提供一种检测替格瑞洛中(1r,2s)-2-(3,4-二氟苯基)环丙胺的方法。本发明提供的一种检测替格瑞洛中(1r,2s)-2-(3,4-二氟苯基)环丙胺的方法,包括以下步骤:a、取(1r,2s)-2-(3,4-二氟苯基)环丙胺(r)-扁桃酸盐,制备对照品溶液,用液相色谱-质谱联用(lc-ms)法进行检测;b、取待检的替格瑞洛样品,制备供试品溶液,用lc-ms法进行检测;c、计算替格瑞洛样品中的(1r,2s)-2-(3,4-二氟苯基)环丙胺含量;步骤a和b所用lc-ms的检测条件为:色谱条件为:用十八烷基硅烷键合硅胶为填充剂;柱温为40℃;以甲酸溶液(1→1000)为流动相a,乙腈为流动相b,进行梯度洗脱;质谱条件为:采用电喷雾电离正离子模式,esi+。进一步的,步骤a中,制备对照品溶液的溶剂为流动相a-乙腈;步骤b中,制备供试品溶液的溶剂为流动相a-乙腈。进一步的,所述的流动相a-乙腈中,流动相a与乙腈的体积比为90:10。进一步的,所述的色谱条件中,十八烷基硅烷键合硅胶为填充剂的色谱为watersxbridgeshieldrp18柱,长度为150mm,内径为4.6mm,填料粒径为3.5μm。进一步的,所述的色谱条件中,梯度程序中0-10分钟流动相b的比例为5%~20%;优选的,流动相b的比例为10%。进一步的,所述的质谱条件中,采用电喷雾电离正离子模式,选择离子检测(sim),定量离子(m/z)为170和/或153。进一步的,所述的质谱条件中,采用电喷雾电离正离子模式,esi+,离子源接口电压为4.5kv;雾化气(氮气)流速为每分钟1.5l;干燥气(氮气)流速为每分钟15l;脱溶剂管温度为350℃;加热块温度为200℃;选择离子检测(sim),定量离子(m/z)为170和/或153;待(1r,2s)-2-(3,4-二氟苯基)环丙胺峰洗脱完毕后将流路切换至废液。在某些实施方案中,本发明提供的一种检测替格瑞洛中(1r,2s)-2-(3,4-二氟苯基)环丙胺的方法,包括以下步骤:a、取(1r,2s)-2-(3,4-二氟苯基)环丙胺(r)-扁桃酸盐,制备对照品溶液,用液相色谱-质谱联用(lc-ms)法进行检测;b、取待检的替格瑞洛样品,制备供试品溶液,用lc-ms法进行检测;c、计算替格瑞洛样品中的(1r,2s)-2-(3,4-二氟苯基)环丙胺含量;步骤a和b所用lc-ms的检测条件为:照高效液相色谱法(中国药典2015年版四部通则0512)测定,用十八烷基硅烷键合硅胶为填充剂;柱温为40℃;以甲酸溶液(1→1000)为流动相a,乙腈为流动相b,按下表进行梯度洗脱;照质谱法(中国药典2015年版四部通则0431)测定,采用电喷雾电离正离子模式,esi+;离子源接口电压为4.5kv;雾化气(氮气)流速为每分钟1.5l;干燥气(氮气)流速为每分钟15l;脱溶剂管温度为350℃;加热块温度为200℃;选择离子检测(sim),定量离子(m/z)为170和/或153;待(1r,2s)-2-(3,4-二氟苯基)环丙胺峰洗脱完毕后将流路切换至废液;进一步的,所述的检测方法,用于(1s,2r)-2-(3,4-二氟苯基)环丙胺的检测。本发明提供了一种检测替格瑞洛中(1r,2s)-2-(3,4-二氟苯基)环丙胺的液相色谱-质谱联用(lc-ms)的检测方法,通过特定的质核比(m/z)进行定性,其他杂质不干扰(1r,2s)-2-(3,4-二氟苯基)环丙胺的检测;同时,本发明方法具有良好的专属性、线性关系、精密度、灵敏度和重复性,加样回收率高,检测结果准确、可靠,为监控替格瑞洛药物中的(1r,2s)-2-(3,4-二氟苯基)环丙胺含量提供了一种行之有效的检测方法,进一步保证了替格瑞洛药物的用药安全。显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。附图说明图1为本发明检测条件下稀释剂的质谱图;图2本发明检测条件下供试品溶液的质谱图;图3为本发明检测条件下对照品溶液的质谱图;图4为对比试验1色谱条件下对照品溶液色谱图;图5为对比试验1色谱条件下供试品溶液的色谱图。具体实施方式本发明具体实施方式中使用的原料、设备均为已知产品,通过购买市售产品获得。替格瑞洛的批号为tcg160505;杂质(1r,2s)-2-(3,4-二氟苯基)环丙胺的对照品(1r,2s)-2-(3,4-二氟苯基)环丙胺(r)-扁桃酸盐的批号为fpm-160405,含量:99.62%;均来源于四川海思科制药有限公司。cpa225d型精密电子天平购自赛多利斯科学仪器有限公司;岛津lcms-2020单四极杆液质联用仪购自岛津公司;岛津lc-20at高效液相色谱仪购自岛津公司;watersxbridgeshieldrp18(4.6mm×150mm,3.5μm)色谱柱购自沃特世公司;agilenttc-c18,4.6mm×150mm,5μm色谱柱购自安捷伦公司。实施例1、本发明检测替格瑞洛中原料杂质的液相色谱-质谱方法仪器:岛津lcms-2020单四极杆液质联用仪;液相色谱条件:色谱柱:watersxbridgeshieldrp18,4.6mm×150mm,3.5μm;流动相:以甲酸溶液(1→1000)为流动相a,乙腈为流动相b,按下表进行梯度洗脱。柱温:40℃;进样量:5μl。质谱条件:采用电喷雾电离正离子模式,esi+;离子源接口电压为4.5kv;雾化气(氮气)流速为每分钟1.5l;干燥气(氮气)流速为每分钟15l;脱溶剂管温度为350℃;加热块温度为200℃。选择离子检测(sim),定量离子(m/z)为170和/或153;待(1r,2s)-2-(3,4-二氟苯基)环丙胺峰洗脱完毕后将流路切换至废液。检测步骤:取替格瑞洛约0.2g,精密称定,用稀释剂[流动相a-乙腈(90:10)]定容至10ml,摇匀,用0.22μm聚四氟乙烯滤膜滤过,取续滤液作为供试品溶液。取(1r,2s)-2-(3,4-二氟苯基)环丙胺(r)-扁桃酸盐对照品适量(乘以0.52,折合为(1r,2s)-2-(3,4-二氟苯基)环丙胺的量),精密称定,加稀释剂[流动相a-乙腈(90:10)]溶解并定量稀释制成每1ml中含1μg的溶液,作为对照品溶液。测定法:取稀释剂及上述溶液各5μl,分别注入液相色谱仪,记录质谱图,结果如图1~图3。图1为实施例1液相-质谱条件下稀释剂的质谱图;图2为实施例1液相-质谱条件下供试品溶液的质谱图,定量离子(m/z)为170与153,结果均未检出。图3为实施例1液相-质谱条件下对照品溶液的质谱图,定量离子(m/z)为170与153,保留时间分别为6.193min与6.200min。结果表明,本发明的液相-质谱条件下,采用专属性强的定量离子进行检测,替格瑞洛及其它杂质不干扰(1r,2s)-2-(3,4-二氟苯基)环丙胺的检测。对比试验1:仪器:岛津lc-20at高效液相色谱仪;色谱柱:agilenttc-c18,4.6mm×150mm,5μm;流动相:以30mmol/l磷酸二氢钾溶液(磷酸调节ph至2.5)为流动相a,乙腈为流动相b,按下表进行梯度洗脱。柱温:35℃;进样量:10μl。检测步骤:取(1r,2s)-2-(3,4-二氟苯基)环丙胺(r)-扁桃酸盐对照品适量,精密称定,加稀释剂[水-乙腈(50:50)]溶解并定量稀释制成每1ml中含0.06mg的溶液,作为对照品溶液。取替格瑞洛样品适量,加稀释剂配置成每1ml约含替格瑞洛1mg的溶液,作为供试品溶液。测定法:上述溶液各10μl,分别注入液相色谱仪,记录色谱图,结果如图4~图5。图4为对比试验1色谱条件下对照品溶液色谱图,扁桃酸与(1r,2s)-2-(3,4-二氟苯基)环丙胺的保留时间分别为13.548min与17.178min;图5为对比试验1色谱条件下供试品溶液的色谱图,存在与(1r,2s)-2-(3,4-二氟苯基)环丙胺保留时间一致的干扰峰。结果表明,采用高效液相色谱法,很难保证测定的专属性,无法进行检测。为了进一步说明本发明的有益效果,本发明提供以下试验例。试验例1、本发明检测方法均采用如下条件:液相色谱条件:色谱柱:watersxbridgeshieldrp18,4.6mm×150mm,3.5μm;流动相:以甲酸溶液(1→1000)为流动相a,乙腈为流动相b,按下表进行梯度洗脱。柱温:40℃;进样量:5μl。质谱条件:采用电喷雾电离正离子模式,esi+;离子源接口电压为4.5kv;雾化气(氮气)流速为每分钟1.5l;干燥气(氮气)流速为每分钟15l;脱溶剂管温度为350℃;加热块温度为200℃。选择离子检测(sim),定量离子(m/z)为170/153;待(1r,2s)-2-(3,4-二氟苯基)环丙胺峰洗脱完毕后将流路切换至废液。1、定量离子:由(1r,2s)-2-(3,4-二氟苯基)环丙胺化学结构与lc-ms原理分析,m/z为153的离子应为(1r,2s)-2-(3,4-二氟苯基)环丙胺脱氨基碎片离子(m-nh2),m/z为170的离子应为准分子离子(m+h),两离子的相对丰度均较高,故拟定采用选择性离子扫描模式(sim),选择m/z为153和/或170的正离子作为(1r,2s)-2-(3,4-二氟苯基)环丙胺的特征离子进行检测。2、专属性实验取(1r,2s)-2-(3,4-二氟苯基)环丙胺(r)-扁桃酸盐对照品适量(乘以0.52,折合为(1r,2s)-2-(3,4-二氟苯基)环丙胺的量),精密称定,加稀释剂[流动相a-乙腈(90:10)]溶解并定量稀释制成每1ml中含1μg的溶液,作为对照品溶液。另取扁桃酸对照品适量,加稀释剂配置成每1ml约含扁桃酸1μg的溶液,作为扁桃酸对照品溶液。另取替格瑞洛样品适量,加稀释剂配置成每1ml约含替格瑞洛1μg的溶液,作为供试品溶液。取稀释剂及上述溶液各5μl,分别注入液相色谱仪,记录质谱图,结果如图1~图4所示。结果表明,本发明的液相-质谱条件下,采用专属性强的定量离子进行检测,扁桃酸与替格瑞洛不干扰(1r,2s)-2-(3,4-二氟苯基)环丙胺的检测。3、定量限和检测限取(1r,2s)-2-(3,4-二氟苯基)环丙胺(r)-扁桃酸盐对照品适量(乘以0.52,折合为(1r,2s)-2-(3,4-二氟苯基)环丙胺的量),精密称定,用稀释剂定量稀释制成一定浓度的溶液。在拟定的液相-质谱条件下进样,记录色谱图,以信噪比(s/n)约为3:1的进样量为检测限,为0.055ng;以信噪比(s/n)约为10:1的进样量为定量限,为0.182ng,满足检测灵敏度及定量的要求。4、线性与范围取(1r,2s)-2-(3,4-二氟苯基)环丙胺(r)-扁桃酸盐对照品适量(乘以0.52,折合为(1r,2s)-2-(3,4-二氟苯基)环丙胺的量),精密称定,用稀释剂定量稀释制成一系列浓度的溶液。分别精密移取5μl,注入液相色谱仪,记录质谱图。结果见表1。表1、线性关系结果表明,(1r,2s)-2-(3,4-二氟苯基)环丙胺在36.48ng/ml~1013.33ng/ml范围内,与峰面积呈良好的线性关系。证明本方法线性范围广,准确度高。5、精密度5.1、重现性取(1r,2s)-2-(3,4-二氟苯基)环丙胺(r)-扁桃酸盐对照品适量(乘以0.52,折合为(1r,2s)-2-(3,4-二氟苯基)环丙胺的量),精密称定,加稀释剂溶解并定量稀释制成每1ml中含0.14μg的溶液,作为对照品溶液。精密吸取对照品溶液5μl,分别注入液质联用色谱仪,连续进样6次,记录质谱图。试验结果见下表2。表2重现性试验结果结果表明,对照品溶液连续进样6次,m/z为170.00与153.00下,(1r,2s)-2-(3,4-二氟苯基)环丙胺峰保留时间的rsd均小于1.0%,峰面积的rsd均小于3.0%。重现性良好。5.2、重复性取替格瑞洛约0.2g,精密称定,用稀释剂[流动相a-乙腈(90:10)]定容至10ml,摇匀,用0.22μm聚四氟乙烯滤膜滤过,取续滤液作为供试品溶液;重复制备6份。另取(1r,2s)-2-(3,4-二氟苯基)环丙胺(r)-扁桃酸盐对照品适量(乘以0.52,折合为(1r,2s)-2-(3,4-二氟苯基)环丙胺的量),精密称定,加稀释剂溶解并定量稀释制成每1ml中含0.14μg的溶液,作为对照品溶液。精密量取对照品溶液与供试品溶液各5μl,分别注入液相色谱-质谱联用仪,记录质谱图。按外标法以峰面积计算。试验结果见下表3。表3重复性试验结果表编号123456含量(%)未检出未检出未检出未检出未检出未检出结果表明,重复测定供试品6次,结果(1r,2s)-2-(3,4-二氟苯基)环丙胺均未检出,重复性良好。6、溶液稳定性取试验例1第5.2项下对照品溶液与供试品溶液,于室温条件下放置,考察6小时内的稳定性。试验结果见下表4。表4溶液稳定性试验结果结果表明,对照品溶液室温放置6小时,m/z为170.00与153.00下,7次测定(1r,2s)-2-(3,4-二氟苯基)环丙胺峰面积的rsd均小于5.0%;供试品中(1r,2s)-2-(3,4-二氟苯基)环丙胺均未检出。说明对照品溶液和供试品溶液在室温下放置6小时稳定。7、耐用性取(1r,2s)-2-(3,4-二氟苯基)环丙胺(r)-扁桃酸盐对照品适量,精密称定,用稀释剂定量稀释制成每1ml中约含1.17μg的溶液,作为对照品储备液;精密量取对照品储备液3.0ml,置25ml量瓶中,用稀释剂稀释至刻度,摇匀,作为对照品溶液。另取替格瑞洛(由试验例1第5.2项下可知(1r,2s)-2-(3,4-二氟苯基)环丙胺未检出)约0.5g,精密称定,置25ml量瓶中,精密加入对照品储备液3ml,加稀释剂稀释至刻度,作为供试品溶液。微调梯度程度初始有机相比例(10±2%)和干燥气流速(15±0.5l/min),精密吸取对照品溶液和供试品溶液各5μl进行测定,记录质谱图,按外标法以峰面积计算各质谱条件下供试品中杂质(1r,2s)-2-(3,4-二氟苯基)环丙胺的回收率变化。试验结果见表5。表5耐用性试验结果色谱-质谱条件m/z=153下回收率(%)m/z=170下回收率(%)已定色谱条件97.7499.40初始有机相比例8%96.43100.54初始有机相比例12%98.7596.70干燥气流速14.5l/min99.0498.97干燥气流速15.5l/min100.2597.66平均(%)98.4498.65rsd(%)1.461.52结果表明,微调梯度程序初始有机相比例(10±2%)和干燥气流速(15±0.5l/min),与原条件相比,m/z为170.00与153.00下,5次测定的平均回收率分别为98.44%与98.65%,rsd分别为1.46%与1.52%。耐用性良好。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1