一种基于多网络图卷积的癌症驱动基因识别方法
1.本发明涉及一种基于多网络图卷积的癌症驱动基因识别方法,属于系统生物学技术领域。
背景技术:
2.少量基因的突变会导致癌症的发生、发展,这样的基因被称为癌症的驱动基因。识别癌症驱动基因对于了解癌症的分子机制、开发精准治疗药物等方向具有重要作用。在过去的十年里,一些大规模的癌症基因组学项目已经公布了来自数千名癌症患者的基因组、基因表达组、转录组和蛋白质组数据。这些项目包括癌症基因组图谱(tcga)、国际癌症基因组联盟(icgc)和癌症体细胞突变目录(cosmic)。这些数以千计的癌症基因组数据促进了识别癌症驱动基因的计算方法的发展。
3.这些计算方法是基于癌症驱动因素的一个或多个特性而设计的。早期的方法只通过使用基因的突变特征,认为越频繁突变的基因越可能是驱动基因,如:mutsigcv。然而,这些方法往往忽略了突变频率较低的驱动基因。由于癌症驱动基因往往互相作用形成蛋白质复合物。研究人员在算法中引入蛋白质相互作用(ppi)网络。如:hotnet2、muffinn等方法通过衡量基因在蛋白质网络中的重要程度来预测驱动基因。为了提高癌症驱动基因的预测,一些模型通过引入其他类型的数据来降低蛋白质网络的不可靠性。例如基因表达谱、亚细胞信息、组织特异性表达谱和基因功能信息等。除了在基因水平上的改变外,癌症也可以通过转录和表观遗传失调的而发生。例如,基因启动子区域的高甲基化或低甲基化可导致关键的肿瘤抑制因子或癌基因活性的沉默,促进癌症生长。
4.生物网络是描述生物实体特征及其关系的有效工具。近年来,人们开发了许多基于生物网络的整合多组学数据的方法。网络表示学习是一种新兴的技术,它在尽可能保留原始网络结构和节点特征的同时,学习所有网络节点的低维特征向量。因此,它可以有效地降低网络中的噪声,提取不同类型的节点特征。一些基于深度神经网络的方法,如deepwalk,node2vec,已被用来学习网络节点的特征。将这些特征与分类器相结合,可以预测癌症驱动基因。图卷积网络(gcn)是一种新的网络表示方法,它可以通过自然地整合网络结构与节点特征来学习网络节点的特征。emogi是一种基于gcn的方法,它利用生物组学数据作为基因特征,并结合蛋白质相互作用网络来学习更复杂的基因特征。但是它没有考虑到基因生物特征本身之间的相似性。
技术实现要素:
5.本发明要解决的技术问题是提供一种基于多网络图卷积的癌症驱动基因识别方法,基于图卷积神经网络,集成多组学数据,能有效提高癌症驱动基因识别的准确性,从而解决现有技术汇中,基于图卷积来预测癌症驱动基因准确度低的问题。
6.本发明的技术方案是:一种基于多网络图卷积的癌症驱动基因识别方法,具体步骤为:
7.step1:根据蛋白质相互作用网络得到结构网络;
8.step2:为每个基因计算出增强特征;
9.step3:根据基因的生物特征之间的相似度得到特征网络;
10.step4:将结构网络、特征网络和生物特征放入多网络图卷积模型(mngcn)中,对模型进行训练,并利用训练好的模型预测新的癌症驱动基因;
11.step5:输出每个基因是否是癌症驱动基因的预测分数。
12.所述step1具体为:删除蛋白质相互作用数据中分数小于0.5的交互数据。
13.所述step2具体为:
14.首先求取基因的差异甲基化率,即一个癌症类型的所有样本中,癌症和匹配正常样本之间甲基化信号差异的平均值:
[0015][0016]
式中,表示基因i在癌症c型中的甲基化值,和是癌症和匹配正常样品中的甲基化信号,sc表示癌症的样本集;
[0017]
然后获取每个基因的差异表达率,每个基因的差异表达率是通过其在癌症中的表达值与正常样本之间的对数倍变化来测量的,然后在所有样本中取平均值;
[0018]
最后,在结构网络中使用deepwalk算法得到每个基因的包含深层关联关系的网络结构特征,确定网络结构特征的长度,然后将其与每个基因在n种癌症中的突变率、差异甲基化率、差异表达率串联在一起,并进行最小最大规范化,得到每个基因的增强特征。
[0019]
基于图神经网络的方法受限于过平滑问题,卷积层的层数往往不宜过多,但较少的层数会使得模型忽略基因在网络中深层的关联关系,而基因在相互作用网络中的多阶邻居信息对提高预测驱动基因的性能很有帮助。受到之前的基于随机游走的方法的启发,本发明在相互作用网络中使用deepwalk算法得到每个基因的包含深层关联关系的网络结构特征,然后通过与生物特征拼接的方式得到增强特征,直接的利用结构信息,提升了预测的准确度。
[0020]
所述step3具体为:根据step2中得到的增强特征,计算基因之间的余弦相似度,得到余弦相似度矩阵,每个基因根据余弦相似度矩阵找到与其最相似的20个邻居用边相连,得到特征网络。
[0021]
余弦相似度矩阵是n*n的矩阵行名和列名都是基因,里面的每一个值就是对应的两个基因的相似度,矩阵中值都处于(0,1)之间,越接近1越相似。本发明根据余弦相似度矩阵找出最大20个值来确定每个基因最相似的20个基因。
[0022]
所述step4中所使用的多网络图卷积模型是基于使用切比雪夫算子的图卷积层,输入为节点特征,以及结构网络和特征网络,将节点特征、结构网络,节点特征、特征网络分别输入两组图卷积,并对经过两组图卷积得到的嵌入矩阵进行一致性约束,最后通过特征拼接、全连接层得到并输出预测分数。
[0023]
本发明的有益效果是:本发明使用了生物多组学数据,并根据蛋白质相互作用网络得到结构网络,根据基因的生物特征之间的相似度得到特征网络,使用多网络图卷积模型来进行癌症驱动基因的预测。
[0024]
本发明的实验结果表明和现有的基于机器学习、图表征学习预测驱动基因的方法相比,本发明提出的方法能提高识别癌症驱动基因的准确性,能为生物学家进行癌症驱动基因识别的实验和进一步研究提供有价值的参考信息。
附图说明
[0025]
图1是本发明mngcn的流程图;
[0026]
图2是本发明mngcn所使用的多网络图卷积模型的结构图。
具体实施方式
[0027]
下面结合附图和具体实施方式,对本发明作进一步说明。
[0028]
实施例1:利用本发明所述的方法对泛癌癌症驱动基因进行预测,蛋白质相互作用数据来自共识路径数据库(cpdb);基因突变,基因甲基化和基因表达的数据来自tcga,涉及8000多个样本和16种不同的癌症类型(肺腺癌、肺鳞癌、肝癌、胃癌、结肠癌、食管癌、直肠癌、头颈癌、肾透明细胞癌、肾乳头状细胞癌、甲状腺癌、子宫内膜癌、宫颈癌、乳腺癌、前列腺癌、膀胱癌)。
[0029]
阳性样本列表是从标有该癌症类型的ncg 6.0中收集的。对于负面样本列表,从所有基因开始,递归地删除ncg,cosmic,omim数据库和kegg癌症通路中的基因。最后,得到的基准数据集包括796个阳性样本和2187个阴性样本。
[0030]
如图1所示,一种基于多网络图卷积的癌症驱动基因识别方法,具体步骤为:
[0031]
step1:根据蛋白质相互作用网络得到结构网络;
[0032]
在删除蛋白质相互作用数据中分数小于0.5的交互后,获得了结构网络。所述结构网络包括13627个节点和504378条边。
[0033]
step2:为每个基因计算出增强特征;
[0034]
基因的突变率被定义为该癌症类型中所有样本的单核苷酸变异(snvs)和拷贝数畸变(cnas)的平均值。
[0035]
基因的差异甲基化率是一个癌症类型的所有样本中,癌症和匹配正常样本之间甲基化信号差异的平均值。定义如下:
[0036][0037]
其中表示基因i在癌症c型中的基因甲基化值。和是癌症和匹配正常样品中的甲基化信号。sc表示癌症的样本集。
[0038]
在一种癌症类型中,每个基因的差异表达率是通过其在癌症中的表达值与正常样本之间的对数倍变化来测量的,然后在所有样本中取平均值。
[0039]
在结构网络中使用deepwalk算法得到每个基因的包含深层关联关系的网络结构特征,长度为16维,然后将其与每个基因在16种癌症中的突变率、差异甲基化率、差异表达率串联在一起,并进行了最小最大规范化。这样,每个基因都得到一个64维的增强特征。
[0040]
step3:根据基因的生物特征之间的相似度得到特征网络;
[0041]
根据步骤2中得到的增强特征,计算基因之间的余弦相似度,得到余弦相似度矩
阵,将每个基因与其最相似的20个邻居用边相连,得到特征网络,包括13627个节点和437920条边。
[0042]
step4:将结构网络、特征网络和生物特征放入多网络图卷积模型(mngcn)中,对模型进行训练,并利用训练好的模型预测新的癌症驱动基因;所述多网络图卷积模型的结构如图2所示。
[0043]
其中切比雪夫图卷积层1、2的设置:输入维度64,输出维度300,切比雪夫滤波器尺寸设置为2,激活函数选择relu函数。切比雪夫图卷积层3、4的设置:输入维度300,输出维度100,切比雪夫滤波器尺寸设置为2,激活函数选择relu函数。全连接层的输入维度为200,输出维度为1。训练过程中的dropout值统一设置为0.5。
[0044]
切比雪夫图卷积层相比于普通的图卷积层能更灵活地提取多阶邻居的特征,具有更强的提取基因特征和传播信息的能力,其具体为:
[0045][0046]z(k)
通过以下定义递归计算:
[0047]z(1)
=x
[0048][0049][0050]z(k)
是一个将切比雪夫多项式带入图神经网络而定义的算子,x是特征矩阵。
[0051]
式中,h表示切比雪夫图卷积层学习的基因特征。θ∈rg×h是神经网络的权重参数矩阵。h是隐藏层维数。k控制切比雪夫滤波器尺寸。此处k=2。表示缩放和标准化的拉普拉斯矩阵,其中i是单位矩阵,l是输入网络的正则化的拉普拉斯矩阵,λ
max
是l的最大特征值。f是激活函数,例如relu。
[0052]
将结构网络、特征网络和生物特征放入多网络图卷积模型。模型对一个基因是否是癌症驱动基因的预测打分可以表示为:
[0053][0054]
其中和是基因i分别通过切比雪夫卷积层3和4得到的嵌入向量,符号代表拼接操作,w和b是可以学习的参数。
[0055]
多网络图卷积模型在训练过程中需要通过最小化来进行优化的损失函数l
total
如下:
[0056]
l
total
=l
pre
+αl
con
[0057]
其中,l
pre
是预测损失,l
con
是一致性约束,α是限制l
con
的超参数,我们将其设置为0.0001。
[0058]
使用交叉熵损失函数来计算预测损失l
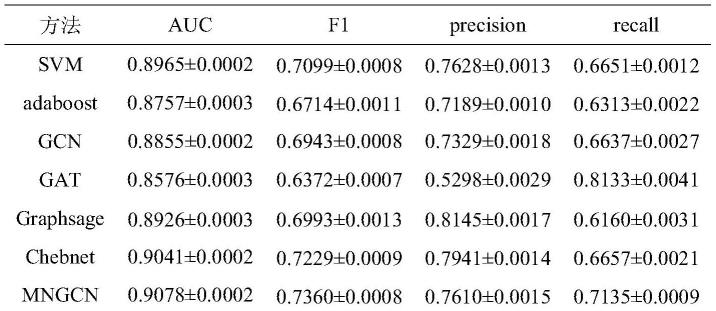
pre
:
[0059]
[0060]
yi表示基因i的已知标签(0或1),n表示训练集中的基因数。θ表示模型的参数。预测损失越小代表模型最终的预测精度越高。
[0061]
为了进一步增强切比雪夫卷积层3和4的输出嵌入矩阵h3和h4的共性,设计一个一致性约束l
con
。
[0062]
首先,使用l2归一化将嵌入矩阵归一化为n3和n4。
[0063]
然后,利用两个归一化矩阵捕获n个节点的相似性s1、s2,具体如下:
[0064][0065][0066]
一致性就表示这两个相似矩阵应该是相似的,这就产生了以下约束:
[0067][0068]
step5:输出每个基因是否是癌症驱动基因的预测分数。预测分数越高,基因是癌症驱动基因的可能性就越高。
[0069]
实施例2:利用本发明所述的方法对乳腺癌癌症驱动基因进行预测,蛋白质相互作用数据来自共识路径数据库(cpdb);基因突变,基因甲基化和基因表达的数据来自tcga。
[0070]
阳性样本列表是从标有该癌症类型的ncg 6.0中收集的。对于负面样本列表,我们从所有基因开始,递归地删除ncg,cosmic,omim数据库和kegg癌症通路中的基因。最后,我们的基准数据集对于;对于乳腺癌包括202个阳性样本和2187个阴性样本。
[0071]
如图1所示,一种基于多网络图卷积的癌症驱动基因识别方法,具体步骤为:
[0072]
step1:根据蛋白质相互作用网络得到结构网络;
[0073]
在删除蛋白质相互作用数据中分数小于0.5的交互后,获得了结构网络。所述结构网络包括13627个节点和504378条边。
[0074]
step2:为每个基因计算出增强特征;
[0075]
基因的突变率被定义为该癌症类型中所有样本的单核苷酸变异(snvs)和拷贝数畸变(cnas)的平均值。
[0076]
基因的差异甲基化率是一个癌症类型的所有样本中,癌症和匹配正常样本之间甲基化信号差异的平均值。定义如下:
[0077][0078]
其中表示基因i在癌症c型中的基因甲基化值。和是癌症和匹配正常样品中的甲基化信号。sc表示癌症的样本集。
[0079]
在一种癌症类型中,每个基因的差异表达率是通过其在癌症中的表达值与正常样本之间的对数倍变化来测量的,然后在所有样本中取平均值。
[0080]
在结构网络中使用deepwalk算法得到每个基因的包含深层关联关系的网络结构特征,长度为16维,然后将其与每个基因在肺腺癌中的突变率、差异甲基化率、差异表达率串联在一起,并进行了最小最大规范化。这样,每个基因都得到一个19维的增强特征。
[0081]
step3:根据基因的生物特征之间的相似度得到特征网络;
[0082]
根据步骤2中得到的增强特征,计算基因之间的余弦相似度,得到余弦相似度矩
阵,将每个基因与其最相似的20个邻居用边相连,得到特征网络,包括13627个节点和437920条边。
[0083]
step4:将结构网络、特征网络和生物特征放入多网络图卷积模型(mngcn)中,对模型进行训练,并利用训练好的模型预测新的癌症驱动基因;所述多网络图卷积模型的结构如图2所示。
[0084]
其中切比雪夫图卷积层1、2的设置:输入维度19,输出维度150,切比雪夫滤波器尺寸设置为2,激活函数选择relu函数。切比雪夫图卷积层3、4的设置:输入维度150,输出维度50,切比雪夫滤波器尺寸设置为2,激活函数选择relu函数。全连接层的输入维度为100,输出维度为1。训练过程中的dropout值统一设置为0.5。
[0085]
切比雪夫图卷积层相比于普通的图卷积层能更灵活地提取多阶邻居的特征,具有更强的提取基因特征和传播信息的能力,其具体为:
[0086][0087]z(k)
通过以下定义递归计算:
[0088]z(1)
=x
[0089][0090][0091]
式中,h表示切比雪夫图卷积层学习的基因特征。θ∈rg×h是神经网络的权重参数矩阵。h是隐藏层维数。k控制切比雪夫滤波器尺寸。此处k=2。表示缩放和标准化的拉普拉斯矩阵,其中i是单位矩阵,l是输入网络的正则化的拉普拉斯矩阵,λ
max
是l的最大特征值。f是激活函数,例如relu。
[0092]
将结构网络、特征网络和生物特征放入多网络图卷积模型。模型对一个基因是否是癌症驱动基因的预测打分可以表示为:
[0093][0094]
其中和是基因i分别通过切比雪夫卷积层3和4得到的嵌入向量,符号代表拼接操作,w和b是可以学习的参数。
[0095]
多网络图卷积模型在训练过程中需要通过最小化来进行优化的损失函数l
total
如下:
[0096]
l
total
=l
pre
+αl
con
[0097]
其中,l
pre
是预测损失,l
con
是一致性约束,α是限制l
con
的超参数,我们将其设置为0.0001。
[0098]
使用交叉熵损失函数来计算预测损失l
pre
:
[0099][0100]
yi表示基因i的已知标签(0或1),n表示训练集中的基因数。θ表示模型的参数。预
测损失越小代表模型最终的预测精度越高。
[0101]
为了进一步增强切比雪夫卷积层3和4的输出嵌入矩阵h3和h4的共性,设计一个一致性约束l
con
。
[0102]
首先,使用l2归一化将嵌入矩阵归一化为n3和n4。
[0103]
然后,利用两个归一化矩阵捕获n个节点的相似性s1、s2,具体如下:
[0104][0105][0106]
一致性就表示这两个相似矩阵应该是相似的,这就产生了以下约束:
[0107][0108]
step5:输出每个基因是否是癌症驱动基因的预测分数。预测分数越高,基因是癌症驱动基因的可能性就越高。
[0109]
基于多网络图卷积的癌症驱动基因识别方法的性能评估:
[0110]
为了评估多网络图卷积(mngcn)的性能,将提出的多网络图卷积方法同以前提出的传统机器学习、图卷积类方法进行了比较。选择auc,f1分数,精准率,召回率作为评价指标,并且做了单网络图卷积与多网络图卷积的比较实验,所有的实验结果都通过十次5倍交叉验证后取平均得到。
[0111]
预测泛癌癌症驱动基因的实验结果如表1所示,预测乳腺癌癌症驱动基因的实验结果如表2所示。
[0112][0113]
表1
[0114][0115]
表2
[0116]
从表1、表2中可以看出,mngcn方法的auc和f1分数指标都比别的预测方法好,精准
率,召回率表现出色。可以看出本发明方法mngcn比起其余现有的方法在预测癌症驱动基因上优势非常明显。
[0117]
基于多网络图卷积的癌症驱动基因识别多网络策略的性能评估:
[0118]
为了验证多网络图卷积(mngcn)的多网络策略能提升性能。只使用结构网络的图卷积模型(mngcn-s)和只使用特征网络的图卷积模型(mngcn-f)与多网络图卷积的比较实验,结果如表3所示。
[0119]
表3
[0120][0121]
从表3中可以看出,本发明的auc、f1分数、召回率指标都比使用单个网络的方法好,证明了多网络图卷积的多网络策略能够提升预测性能。
[0122]
综上所述,在与其他预测方法比较之后,证明了基于多网络图卷积的癌症驱动基因识别方法的有效性。
[0123]
以上结合附图对本发明的具体实施方式作了详细说明,但是本发明并不限于上述实施方式,在本领域普通技术人员所具备的知识范围内,还可以在不脱离本发明宗旨的前提下作出各种变化。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1