用于确定双基因或寡基因变异的组合的致病性的预测方法与流程
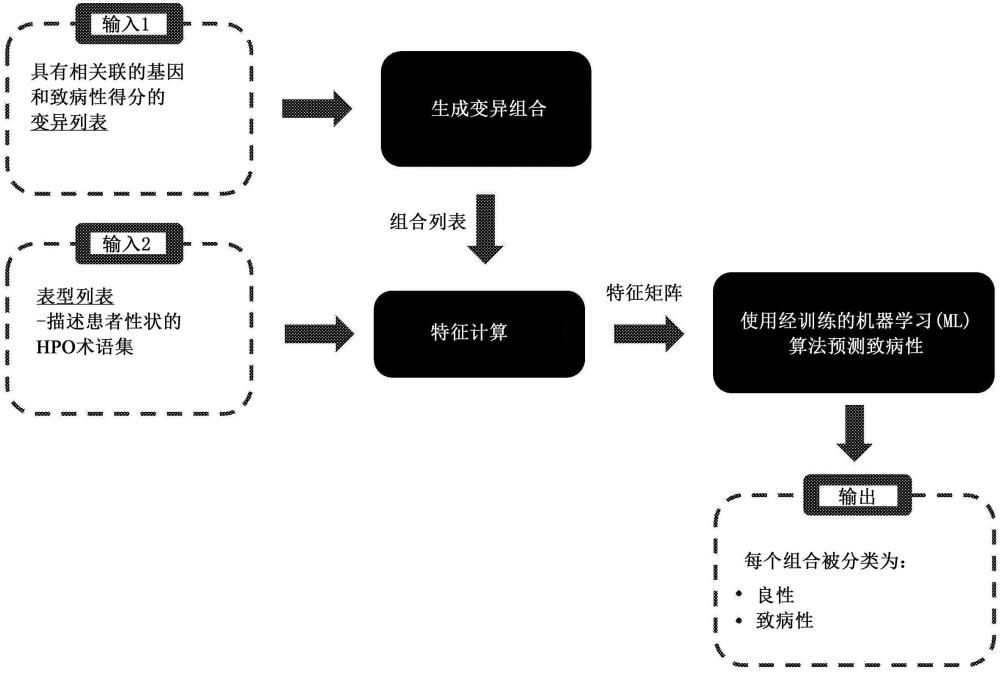
本发明涉及一种用于确定双基因或寡基因变异的组合的致病性的预测方法。因此,本发明的一般是在基因组学和/或医学遗传研究背景下使用的通过电子计算执行的预测方法,以支持预测预后。
背景技术:
1、几十年来,遗传病的遗传机制一直通过还原范式“一个基因,一种疾病”来解释,根据该范式,损害单个基因的突变(也称为变异)被证明是几种孟德尔遗传的罕见疾病的病因。
2、近年来已经开发了更复杂的遗传模型来解释一系列遗传疾病,这些疾病的病因不能通过单个基因的突变来解决(例如,参见katsanis n.“the continuum of causality inhuman genetic disorders(人类遗传疾病的因果的连续性)”.genome biol.2016;17(1):233.https://doi:10.1186/s13059-016-1107-9)。
3、这些病理性疾病中的一些需要多个同时发生突变的基因来出现疾病的症状,而其他的病理性疾病的特征在于主要致病基因和边界基因网络,边界基因网络的作用是调节疾病的结果、症状的严重程度或其发作。
4、最近,已经开发了一些基于机器学习的模型来将双基因变异的组合区分为致病或良性变异。在这方面,应参考例如下列科学论文:
5、papadimitriou s.,gazzo a.,versbraegen n.,nachtegael c.,aerts j.,moreau y.,van dooren s.,nowe a.,smits g.,lenaerts t.,“predicting disease-causing variant combinations(预测致病变异组合)”.proc.natl.acad.sci.u.s.a.2019;116(24),11878-11887.https://doi.org/10.1073/pnas.1815601116
6、mukherjee s.,cogan j.,newman j.,phillips j.,hamid r.,meilerìj.capra,j.,“identifying digenic disease genes using machine learning in theundiagnosed diseases network(在未诊断疾病网络中使用机器学习识别双基因疾病基因)”,preprint at biorxiv.2020.05.31.125716.
7、doi:https://doi.org/10.1101/2020.05.31.125716
8、renaux a.,papadimitriou s.,versbraegen n.,nachtegael c.,boutry s.,nowéa.,smits g.,lenaerts t.“orval:a novel platform for the prediction andexploration of disease-causing oligogenic variant combinations(用于预测和探索致病寡基因变异组合的新平台)”.nucleic acids res.2019jul2;47(w1):w93-w98.https://doi:10.1093/nar/gkz437.
9、然而,这些方法用分子、病理或预测效应信息分别丰富每个变异,并整合了双基因相互作用信息,寻找两个相关基因共有的进化模式和生物学途径。
10、这些方法的主要限制在于完全没有考虑与患者的表型相关的信息,因此忽略了可以为准确识别致病变异提供非常有用的信息的方面。
技术实现思路
1、本发明的目的是提供一种通过计算机处理实现的预测方法,以确定双基因或寡基因变异的组合的致病性,该方法允许至少部分地解决上文参考现有技术描述的缺点,并响应在所考虑的技术领域中特别感觉到的上述需求。
2、这一目的通过根据权利要求1的方法来实现。
3、这种方法的进一步实施方式由权利要求2-18限定。
技术特征:
1.一种确定与疾病相关的双基因或寡基因变异的组合的致病性的计算机实现的方法,包括以下步骤:
2.根据权利要求1所述的方法,适于确定双基因变异的致病性,其中:
3.根据前述权利要求中任一项所述的方法,其中,描述所述患者的表型性状的步骤包括:
4.根据权利要求3所述的方法,其中,hpo对表型性状的描述由有向无环图表示。
5.根据前述权利要求中任一项所述的方法,在对于所述患者中识别的每个变异计算致病性指数或得分的步骤之后并且在描述患者的表型性状的步骤之后,所述方法还包括以下步骤:
6.根据前述权利要求中任一项所述的方法,其中,对于所考虑的每个基因,所述基因-表型关联特征包括:
7.根据前述权利要求中任一项所述的方法,其中,对于每个基因组合,所述双基因性或寡基因性特征包括:
8.根据前述权利要求中任一项所述的方法,其中,对于每个基因,所述基因的所述先验属性特征包括:
9.根据前述权利要求中任一项所述的方法,在使用所述经训练的算法之前,所述方法包括进一步的初步训练步骤,所述初步训练步骤基于包含涉及已知病例的数据的所述训练数据集的两个子集来执行,
10.根据权利要求8所述的方法,其中,初步训练步骤包括训练多个经训练的算法或分类器,并且评估每个分类器的性能。
11.根据权利要求10所述的方法,还包括基于所述初步性能评估,选择用于处理的经训练的算法的子集的步骤。
12.根据权利要求9或权利要求11所述的方法,其中,一旦完成了所述初步训练步骤,则处理用于所述致病性确定的所述输入信息的步骤包括:通过训练中使用的所有经训练的算法或分类器或者通过在训练期间选择的所述经训练的算法或分类器来处理信息。
13.根据权利要求10-12中任一项所述的方法,其中,所述经训练的算法或分类器包括以下中的一项或多项:
14.根据权利要求9-13中任一项所述的方法,其中,所述第一训练子集或“训练集”从两个数据源生成:
15.根据权利要求14所述的方法,还包括在数据资源朝向阴性病例不平衡的情况下平衡数据,以获得两个类别之间的平衡分布的步骤,
16.根据前述权利要求中任一项所述的方法,其中,所述输出信息包括所考虑的双基因或寡基因变异的至少一种组合、或者所考虑的双基因或寡基因变异中的多个变异组合、或者所考虑的双基因或寡基因变异的所有组合的估计致病概率。
17.根据权利要求16所述的方法,其中,对于双基因或寡基因变异的每种组合,所述输出信息还包括表示双基因或寡基因变异的所述组合是“致病的”还是“良性的”的二元结果,所述二元结果通过将为双基因或寡基因变异估计的致病概率与双基因或寡基因变异的所述组合本身所关联的相应阈值进行比较而获得。
18.根据前述权利要求中任一项所述的方法,其中,所述输出信息包括从所考虑的寡基因或双基因变异的组合集中识别出变异的最相关寡基因组合或变异的最佳双基因对,所述最相关寡基因组合或所述最佳双基因对与描述所述患者的表型集具有最相关的相关性。
技术总结
描述了一种确定与疾病相关的双基因或寡基因变异的组合的致病性的计算机实现的方法。该方法首先包括定义必须确定致病性的变异集。这种变异指存在于至少两个基因中的相应基因的一个或两个等位基因中的突变,其中,每个基因与两个相应的等位基因相关联。该方法然后包括关于所考虑的至少两个基因的等位基因中存在或不存在前述变异,确定可能发生的情况的步骤。每种情况与相应的组合相关联,在组合中,每个变异存在于所考虑的所有基因的所有可能的等位基因子集中的相应等位基因子集中,或者该变异存在于所考虑的所有基因的所有等位基因中。对于每一种定义的情况,即对于每种组合和每个基因,该方法然后包括计算致病性指数或得分,其适于估计一个或多个相应变异在多大程度上改变相应基因的功能。该方法还包括以下步骤:通过标准化的表型术语,即适于描述在患者中发现的表型异常的标准化信息,描述患者的表型性状,并计算或准备用于致病性确定的输入信息。这种信息包括以下四种类型的特征:基因‑表型关联特征,针对所考虑的每个基因单独计算,并适于衡量患者的上述表型性状与已知与单个基因关联的表型有多少重叠;双基因性或寡基因性特征,针对每个上述基因组合计算,并适于捕获形成每种组合的基因之间的相互作用;基因的先验属性特征,针对所考虑的每个上述基因计算;变异相关特征,基于与所考虑的所有组合相关的上述致病性指数或得分,针对所考虑的每个基因而计算。该方法还包括将用于致病性确定的所述输入信息提供给至少一个经训练的算法,并通过所述至少一个经训练的算法处理用于致病性确定的所述输入信息。经训练的算法是由人工智能和/或机器学习技术训练的算法。基于已知病例的训练数据集,在初步训练步骤中训练该算法,将为每个已知病例计算的上述输入信息提供给待训练的算法,并基于相应已知病例的致病性/良性的知识训练该算法。最后,该方法包括从经训练的算法获得输出信息的步骤,该输出信息表示所考虑的变异或突变的每种组合的致病性。
技术研发人员:伊万·利蒙杰利,苏珊娜·祖卡,费代丽卡·德·保利,埃托雷·里佐,保罗·马尼,费代丽卡·巴卡利尼
受保护的技术使用者:基因组有限责任公司
技术研发日:
技术公布日:2024/1/25
- 还没有人留言评论。精彩留言会获得点赞!