一种分子动力学的快速建模方法与程序库
本发明涉及耗散粒子动力学,具体涉及一种分子动力学的快速建模方法与程序库。
背景技术:
1、耗散粒子动力学(dissipative particle dynamics,dpd)是一种基于分子动力学的模拟方法,适用于研究流体和软物质体系,通常采用lammps软件进行模型。对于lammps模拟所需的初始模型文件要求详细的阐述初始体系中的原子信息,例如原子的种类、质量,以及哪几个原子间成键、哪几个原子间成角,并且需要给定每个粒子的初始坐标位置。
2、在传统技术方案中,一种方式是采用vmd一类的建模软件进行建模,但是存在功能固定,操作复杂,学习成本较高的问题。尤其是对于大型分子、复杂的体系或者含有非常规化合物的体系,建模极其困难甚至是无法实现,难以完全符合构想的体系模型。另一种方式是使用c语言、fortran、python等语言,通过自定义脚本生成原子类型与模型位置,但是使用自定义脚本生成初始的原子类型和位置模型需要具有一定的编程经验和能力,对于不熟悉编程的用户可能会有一定的困难。同时,对于复杂的体系,例如含有大量原子或者非常规的结构,手动编写脚本并生成初始模型会变得非常困难,且存在程序脚本与建模体系高绑定,难以迁移的问题,无法在已有的模型文件基础上进行进一步的修改。同时在使用脚本文件编写复杂体系模型时,往往会出现脚本语法错误或者是编写的逻辑错误,导致生成的初始模型不符合实际情况,且代码难以阅读,不利于后期修改。
技术实现思路
1、针对现有技术存在的不足,本发明的目的在于提供一种分子动力学的快速建模方法与程序库,该建模方法简单、易懂、快速、学习成本低并且可以完成复杂体系的模型构建。
2、为实现上述目的,本发明提供了如下技术方案:一种分子动力学的快速建模方法,包括分子模型和系统模型,所述系统模型由分子模型组成,且系统模型是一组分子模型的集合或是多个子系统合并成的最终体系模型。
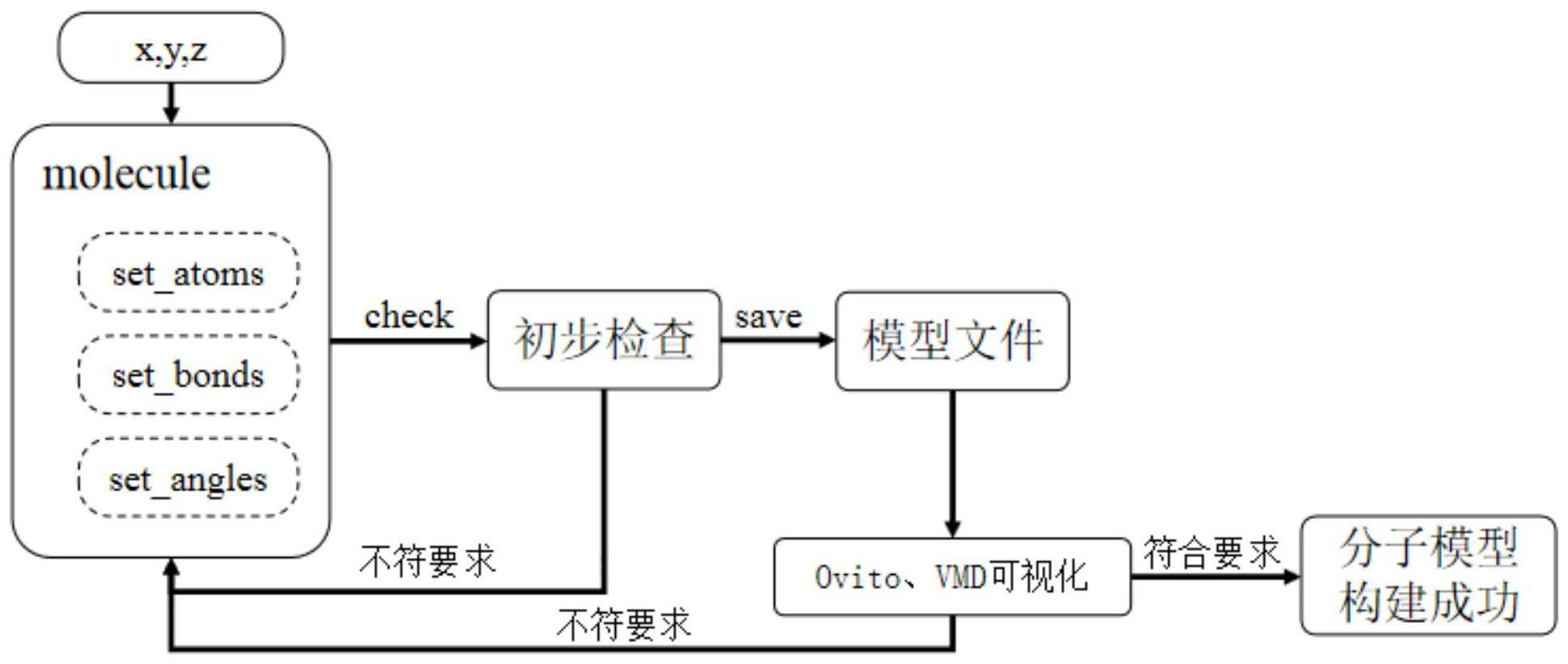
3、作为优选的:在分子模型构建中,定义molecule数据类型,将建模文件所需的原子类型与坐标信息atoms、原子间成键信息bonds和原子间成角信息angles三个作为molecule数据类型基本数据结构;其中atoms数据包括原子序号id、原子类型type和原子坐标xyz;bonds数据包括原子序号id、原子类型type和原子坐标xyz;bonds数据包括成键序号、成键类型type和两个成键粒子的id;angles数据包括成角序号、成角类型type和三个成键粒子的id。
4、作为优选的:在构建时,先使用定义原子的xyz坐标信息,通过调用函数类型molecule(xyz)获得分子模型,根据每个粒子的坐标先后顺序,对粒子由1开始依次排序,自动生成粒子序号,随后通过molecule.set_atoms指令批量设置原子id所对应的原子类型,通过molecule.set_bonds指令批量设置原子间成键与成键类型,通过molecule.set_angles指令批量设置原子间成角与成角类型,并且通过中括号与括号两种方式分别表示单独成键角以及连续成键角,通过重载+方法,实现多个分子模型间的合并,或者使用molecule.append指令追加已有的分子模型,并且可以通过molecule.space_change指令实现分子坐标修改或者是进行整体偏移,通过molecule.space指令返回原有的各原子坐标,定义molecule.to_material指令实现molecule数据类型到material数据类型的切换。
5、作为优选的:系统模型搭建,定义material数据结构,在分子模型数据结构atoms中多一个组group的数据。
6、作为优选的:分子模型构建后,通过material.append指令将各分子单元加入系统模型中,将molecule类型加入系统中时,可以指定数据group的值,默认是在系统中已有的group个数上加一,其余atoms、bonds和angles的序号与原子id偏移问题会自动重新对应;并且通过material.space_change指令实现系统的坐标修改或者是进行整体偏移;通过material.space指令返回原有的各原子坐标;通过material.loc_id指令实现提取出指定原子id所对应的atoms、bonds和angles值为新material数据类型;通过material.loc_type指令实现提取出指定原子类型type、键类型type和角类型type所对应的所有原子id数据的atoms、bonds和angles值,并返回为新material数据类型;material.loc_group三个指令实现提出指定的组所包括的atoms、bonds和angles值;同时,通过重载+方法,实现多个系统体现间的合并,系统中atoms、bonds和angles值所对应的原子id偏移问题会自动重新对应;定义方法load指令实现导入已有的模型文件数据,并返回成material数据类型;定义material.to_molecule指令实现material数据类型到molecule数据类型的切换。
7、作为优选的:定义通用的方法check检查构建的molecule或material数据类型是否存在atoms、bonds和angles值异常,包含原子类型是否全部定义检测,bonds与angles中是否存在未定义的原子id检测,以及atoms、bonds和angles的原子类型值不连续检测,并返回异常数据的id与异常类型;若检测正常,则输出该模型中现有的粒子个数总数、键个数总数、角个数总数以及体系所占用的空间位置。
8、作为优选的:定义通用方法save实现molecule或material数据类型的导出,保存为可被lammps、ovito、vmd识别的通用模型文件,并且可以通过ovito或者是vmd进行可视化观察。
9、作为优选的:模型可以从已有的建模文件中导入。也可以完全由自己从头构建,通过调用相应的指令在数据类型molecule与material中增删相应数据,即可完成自定义模型构建。
10、本发明还将提供一种分子动力学的程序库,该程序库包括上述的快速建模方法,即包括分子模型和系统模型,所述系统模型由分子模型组成,且系统模型是一组分子模型的集合或是多个子系统合并成的最终体系模型。
11、采用上述技术方案,本发明具有如下有益效果:
12、本发明将快速建模过程分为分子模型构建与系统模型搭建两个部分,简单易懂的快速建模方法,在基于python的基础上提供一种新的快速建模库,学习成本低,并且可以完成复杂体系的模型构建,实用性好。
13、下面结合说明书附图和具体实施例对本发明作进一步说明。
技术特征:
1.一种分子动力学的快速建模方法,其特征在于:包括分子模型和系统模型,所述系统模型由分子模型组成,且系统模型是一组分子模型的集合或是多个子系统合并成的最终体系模型。
2.根据权利要求1所述的一种分子动力学的快速建模方法,其特征在于:在分子模型构建中,定义molecule数据类型,将建模文件所需的原子类型与坐标信息atoms、原子间成键信息bonds和原子间成角信息angles三个作为molecule数据类型基本数据结构;其中atoms数据包括原子序号id、原子类型type和原子坐标xyz;bonds数据包括原子序号id、原子类型type和原子坐标xyz;bonds数据包括成键序号、成键类型type和两个成键粒子的id;angles数据包括成角序号、成角类型type和三个成键粒子的id。
3.根据权利要求2所述的一种分子动力学的快速建模方法,其特征在于:在构建时,先使用定义原子的xyz坐标信息,通过调用函数类型molecule(xyz)获得分子模型,根据每个粒子的坐标先后顺序,对粒子由1开始依次排序,自动生成粒子序号,随后通过molecule.set_atoms指令批量设置原子id所对应的原子类型,通过molecule.set_bonds指令批量设置原子间成键与成键类型,通过molecule.set_angles指令批量设置原子间成角与成角类型,并且通过中括号与括号两种方式分别表示单独成键角以及连续成键角,通过重载+方法,实现分子模型间的合并,或者使用molecule.append指令追加已有的分子模型,并且可以通过molecule.space_change指令实现分子坐标修改或者是进行整体偏移,通过molecule.space指令返回原有的各原子坐标,定义molecule.to_material指令实现molecule数据类型到material数据类型的切换。
4.根据权利要求3所述的一种分子动力学的快速建模方法,其特征在于:系统模型搭建,定义material数据结构,在分子模型数据结构atoms中多一个组group的数据。
5.根据权利要求4所述的一种分子动力学的快速建模方法,其特征在于:在分子模型构建后,通过material.append指令将各分子单元加入系统模型中,将molecule类型加入系统中时,可以指定数据group的值,默认是在系统中已有的group个数上加一,其余atoms、bonds和angles的序号与原子id偏移问题会自动重新对应;并且通过material.space_change指令实现系统的坐标修改或者是进行整体偏移;通过material.space指令返回原有的各原子坐标;通过material.loc_id指令实现提取出指定原子id所对应的atoms、bonds和angles值为新material数据类型;通过material.loc_type指令实现提取出指定原子类型type、键类型type和角类型type所对应的所有原子id数据的atoms、bonds和angles值,并返回为新material数据类型;material.loc_group三个指令实现提出指定的组所包括的atoms、bonds和angles值;同时,通过重载+方法,实现多个系统间的合并,系统中atoms、bonds和angles值所对应的原子id偏移问题会自动重新对应;定义方法load指令实现导入已有的模型文件数据,并返回成material数据类型;定义material.to_molecule指令实现material数据类型到molecule数据类型的切换。
6.根据权利要求5所述的一种分子动力学的快速建模方法,其特征在于:定义通用的方法check检查构建的molecule或material数据类型是否存在atoms、bonds和angles值异常,包含原子类型是否全部定义检测,bonds与angles中是否存在未定义的原子id检测,以及atoms、bonds和angles的原子类型值不连续检测,并返回异常数据的id与异常类型;若检测正常,则输出该模型中现有的粒子个数总数、键个数总数、角个数总数以及体系所占用的空间位置。
7.根据权利要求6所述的一种分子动力学的快速建模方法,其特征在于:定义通用方法save实现molecule或material数据类型的导出,保存为可被lammps、ovito、vmd识别的通用模型文件,并且可以通过ovito或者是vmd进行可视化观察。
8.根据权利要求1或7所述的一种分子动力学的快速建模方法,其特征在于:模型可以从已有的建模文件中导入;也可以完全由自己从头构建,通过调用相应的指令在数据类型molecule与material中增删相应数据,即可完成自定义模型构建。
9.一种分子动力学的程序库,其特征在于:包括权利要求1-8任一权利要求所述的快速建模方法,包括分子模型和系统模型,所述系统模型由分子模型组成,且系统模型是一组分子模型的集合或是多个子系统合并成的最终体系模型。
技术总结
本发明专利公开了一种分子动力学的快速建模方法,包括分子模型和系统模型建模,所述系统模型由分子模型组成,且系统模型是一组分子模型的集合或是多个子系统合并成的最终体系模型。上述技术方案,使用者仅需掌握几条基础命令,实现快速建模,学习成本低。在大型复杂系统建模时,可以通过多个子系统组合的方式进行超大规模的系统构建。通过对生成的模型文件进行快速的增删改查,实现模型的再利用与快速建模,极大的增强了模型的迁移能力。通过对于模型粒子数、边界位置、原子成键角个数的快速统计,实现了建模后的简易检查。在程序脚本建模的基础上,实现了在建模过程中与可视化软件的快速联动,在建模过程中可以通过可视化观察模型的建立是否符合预期。
技术研发人员:牟轩慷,李士本
受保护的技术使用者:温州大学
技术研发日:
技术公布日:2024/1/12
- 还没有人留言评论。精彩留言会获得点赞!