针对人类内源性逆转录病毒的注释方法
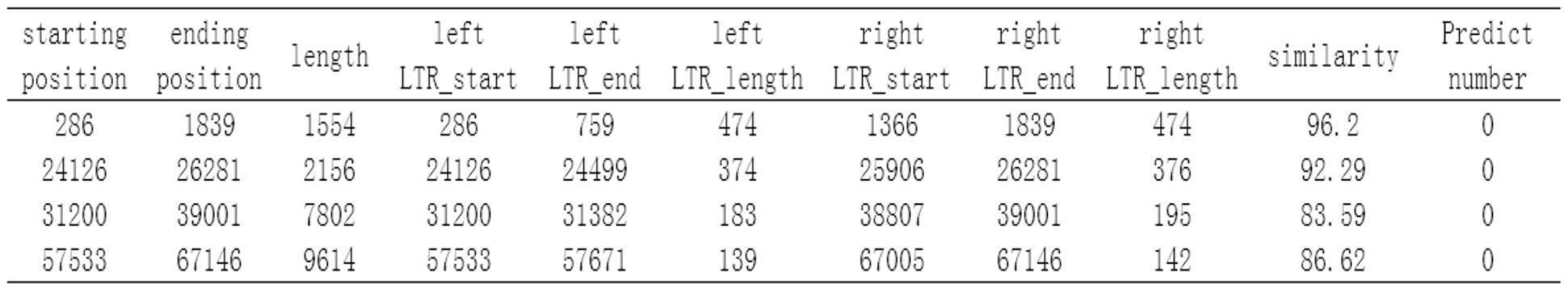
本发明属于生物信息学及基因组研究,尤其涉及一种针对人类内源性逆转录病毒的注释方法。
背景技术:
1、内源性逆转录病毒(erv),也称为ltr转座元件,属于逆转录转座元件的一种,逆转录转座元件是根据两端是否具有长末端重复序列(ltr)分为ltr和非ltr转座元件。erv占到人类基因组8%,在宿主进化的过程中整合,同时可以作为顺式调控元件在基因组中发挥作用。完整的erv由两端序列完全一致的末端重复、衣壳蛋白(gag)和pol构成,后生动物中还包含包膜蛋白(env)。其中pol包含ap(天冬氨酸酶)、int(整合酶)、rt(逆转录酶)和rh(核糖核酸酶h),是erv能否自主转座的关键蛋白。erv和肿瘤或疾病的发生发展、基因表达、物种进化过程高度相关,插入到关键基因内或周围会导致性状的改变,同时诱导的表观遗传变化经常影响相邻基因的差异表达并产生新的调控模式。对erv的研究有助于理解发育和形态进化的多样性,同时其在肿瘤和自身免疫病等领域也有一定程度的应用。近年来,由于erv可能构成合适的生物标志物和疾病治理靶点,已经成为生物信息学及基因组等相关研究领域的热点。
2、目前,不同的物种中erv存在多态性差异,erv对宿主基因调控的程度还尚不清楚,整合进宿主基因的erv及元件因高度变异导致完整度被破坏,erv及其元件的鉴定和注释一直是一大难点,不同方法导致鉴定结果差异很大,结果的解读复杂。再者,不同灵长类物种的erv元件鉴定目前并没有完整的数据库可供搜索,导致相关研究的计算复杂繁琐、耗时费力、操作量大。
3、中国专利“一种用于内源性逆转录病毒的鉴定注释方法”(专利号2022100197828公开日2022年3月25日)中,其erv鉴定注释方法首先选取的病毒蛋白作为探针,识别相似的病毒蛋白序列后向蛋白两侧延伸侧翼序列,由此利用ltrharvest软件,基于隐式马尔科夫模型搜索宿主中的成对ltr序列,进而提取erv病毒候选序列,同时注释各病毒蛋白结构域的结构、位置信息等。在erv的定义中,erv序列是由两侧的ltr序列与ltr序列之间的蛋白结构域(包括gag、pol、pro、env)组成,只含一侧ltr(sololtr)、具有至少一侧ltr同时含至少一个蛋白编码序列都可称为erv。erv分类的基础是根据有无ltr来区分序列是否为erv,同时根据ltr序列的结构的不同来对erv进行亚分类。
技术实现思路
1、本发明要解决的技术问题是提供一种设计合理、方便高效的针对人类内源性逆转录病毒的注释方法。
2、为解决上述技术问题,本发明采用以下技术方案:
3、针对人类内源性逆转录病毒的注释方法,从erv的两侧ltr序列出发,基于隐式马尔科夫模型利用ltrharvest软件首先识别人类全基因组的ltr序列,同时搜集erv相关的蛋白质条目,再利用ltrdigest软件注释基因组erv。
4、上述针对人类内源性逆转录病毒的注释方法,包括以下步骤:
5、<1>获取人类的全基因组数据文件;
6、<2>使用ltr harvest软件对步骤<1>获取的所有全基因组文件进行从头搜索,鉴定人类全基因组序列中的候选内源性逆转录病毒(erv)的两端ltr;
7、<3>构建人类erv相关蛋白质条目的文库;
8、<4>利用ltr digest软件对步骤<3>的蛋白质条目进行整理注释,获取ltr位点、染色体信息以及序列文件,同时自动注释人类erv内部特征。
9、步骤<1>按以下进行:通过ncbi数据库入口,以人类的拉丁名“homo”作为关键词进行搜索,下载所需的全基因组文件。
10、步骤<2>中按以下参数设定进行:指定候选ltr序列和参考序列的相似阈值为80%,相似度低于80%的序列则不作为候选序列;指定ltr候选序列的两端ltr长度范围在1kb-15kb之间,同时输出的结果序列只选择相似性最高的;精确搜索ltr的起始和结尾部位的基序(motif)中的4个核苷酸:tgca,同时不允许错配的核苷酸出现;输出检测目标重复序列(tsd),设定其最小长度为5,最大长度为20,以标准文件形式输出;结果数据根据序列编号对应上染色体,获取序列的起始位置和终止位置,将结果保存为统一的格式。
11、步骤<3>按以下进行:在pfam数据库中使用以“retro”作为关键词,同时结合文献,获取与erv相关的蛋白质相关条目,将蛋白质条目转换问hmmer2格式,整合形成文库。
12、步骤<4>按以下进行:利用局部对比的方法,使用ltr digest软件对候选的ltr内部特征进行注释,确定ltr序列及其内部的长度、位置、方向、距离和序列组成等特征,并计算长度、一致性、覆盖度和重复次数等,输出结果文件。
13、步骤<4>中识别ltr之间的蛋白质结构包括pol、env、gag以及搜索确认的蛋白质条目,同时注释位置。
14、步骤<4>输出结果文件中,手动过滤没有任何命中蛋白质结构的候选erv序列,排除非候选erv序列。
15、针对目前erv鉴定注释存在的问题,发明人建立了一种针对人类内源性逆转录病毒的注释方法,从erv的两侧ltr序列出发,参考其他已发表文献,基于隐式马尔科夫模型利用ltrharvest软件首先识别人类全基因组的ltr序列,同时尽可能搜集erv相关的蛋白质条目,除了gag、pol、pro、env四种erv基础的蛋白结构域之外,再利用ltrdigest软件注释基因组erv。与现有技术相比,本发明通过方法的构建,采用先识别内源性病毒两端的特征序列ltr,接着由此出发鉴定ltr之间的各编码序列及原件等,从而实现高效、快速地对人类全基因组进行内源性逆转录病毒及元件的挖掘、鉴定和注释,增大检出效能,结果更加全面,获得一个注释丰富完善的人类erv特征的数据库,为其结构、定位、变异和进化等研究提供依据;同时,本发明收集并利用了已知的erv相关蛋白其注释更全面,在病原体等相关领域应用广泛。该方法利用人类的内源性逆转录病毒数据,包括了完整的内源性逆转录病毒区域以及区域中病毒的各类元件,从而极大地减少了相关科研工作者的数据计算分析时间,有助于内源性逆转录病毒的溯源、遗传进化的研究,有效解决人类erv及元件的注释方法繁琐复杂的问题,为病原体溯源提供重要依据。应用本发明可以推测内源性逆转录病毒和人类宿主间的进化关系和疾病,进一步分析病毒的多样性和分类问题,推动人类内源性逆转录病毒及相关学科的研究发展。
技术特征:
1.一种针对人类内源性逆转录病毒的注释方法,其特征在于:从erv的两侧ltr序列出发,基于隐式马尔科夫模型利用ltrharvest软件首先识别人类全基因组的ltr序列,同时搜集erv相关的蛋白质条目,再利用ltrdigest软件注释基因组erv。
2.根据权利要求1所述的针对人类内源性逆转录病毒的注释方法,其特征在于包括以下步骤:
3.根据权利要求2所述的针对人类内源性逆转录病毒的注释方法,其特征在于步骤<1>按以下进行:通过ncbi数据库入口,以人类的拉丁名“homo”作为关键词进行搜索,下载所需的全基因组文件。
4.根据权利要求2所述的针对人类内源性逆转录病毒的注释方法,其特征在于步骤<2>中按以下参数设定进行:指定候选ltr序列和参考序列的相似阈值为80%;指定ltr候选序列的两端ltr长度范围在1kb-15kb之间,同时输出的结果序列只选择相似性最高的;精确搜索ltr的起始和结尾部位的基序motif中的4个核苷酸:tgca,同时不允许错配的核苷酸出现;输出检测目标重复序列tsd,设定其最小长度为5,最大长度为20,以标准文件形式输出;结果数据根据序列编号对应上染色体,获取序列的起始位置和终止位置,将结果保存为统一的格式。
5.根据权利要求2所述的针对人类内源性逆转录病毒的注释方法,其特征在于步骤<3>按以下进行:在pfam数据库中使用以“retro”作为关键词,同时结合文献,获取与erv相关的蛋白质相关条目,将蛋白质条目转换问hmmer2格式,整合形成文库。
6.根据权利要求2所述的针对人类内源性逆转录病毒的注释方法,其特征在于步骤<4>按以下进行:利用局部对比的方法,使用ltr digest软件对候选的ltr内部特征进行注释,确定ltr序列及其内部的长度、位置、方向、距离和序列组成等特征,并计算长度、一致性、覆盖度和重复次数等,输出结果文件。
7.根据权利要求6所述的针对人类内源性逆转录病毒的注释方法,其特征在于步骤<4>中识别ltr之间的蛋白质结构包括pol、env、gag以及搜索确认的蛋白质条目,同时注释位置。
8.根据权利要求6所述的针对人类内源性逆转录病毒的注释方法,其特征在于步骤<4>
技术总结
本发明公开了一种针对人类内源性逆转录病毒的注释方法,从ERV的两侧LTR序列出发,基于隐式马尔科夫模型利用LTRharvest软件首先识别人类全基因组的LTR序列,同时尽可能搜集ERV相关的蛋白质条目,再利用LTRdigest软件注释基因组ERV。与现有技术相比,本发明通过方法的构建,采用先识别内源性病毒两端的特征序列LTR,接着由此出发鉴定LTR之间的各编码序列及原件等,从而实现高效、快速地对人类全基因组进行内源性逆转录病毒及元件的挖掘、鉴定和注释,增大检出效能,结果更加全面,获得一个注释丰富完善的人类ERV特征的数据库,为其结构、定位、变异和进化等研究提供依据;同时,本发明收集并利用了已知的ERV相关蛋白其注释更全面,在病原体等相关领域应用广泛。
技术研发人员:韦彗琳,胡艳玲,李兰香,张蝶,蒋春兰,梁柏莹,张玉銮
受保护的技术使用者:广西医科大学
技术研发日:
技术公布日:2024/1/16
- 还没有人留言评论。精彩留言会获得点赞!