幽门螺杆菌对多药耐药表型的预测模型构建方法及装置
本申请涉及耐药性预测,具体涉及一种幽门螺杆菌对多药耐药表型的预测模型构建方法及装置。
背景技术:
1、幽门螺杆菌(helicobacterpylori,h.pylori)感染是世界上最常见的慢性细菌感染之一,感染了全球一半以上的人口。1994年世界卫生组织将它定义为i类致癌因子。2017年,世界卫生组织将耐克拉霉素的h.pylori列类为“高优先级”细菌,2022年美国卫生和公众服务部将h.pylori列为明确致癌物。多项随机对照研究和队列研究表明,根除h.pylori感染可以降低胃癌的发展的风险,是预防胃癌最主要的可控危险因素。
2、在临床实践中,只有少数抗生素例如甲硝唑(mtz)、克拉霉素(clr)、左氧氟沙星(lev)、阿莫西林(amx)和四环素(tet)等被证明对根除h.pylori有效。在过去的40年里利用这些常用药物,人们提出了多种不同组合的治疗方案,然而全球抗生素耐药性回顾分析和荟萃分析显示,h.pylori根除率在逐年下降,多耐药率逐年上升。因此,在使用抗生素之前了解h.pylori药物敏感性状况,进行药敏指导下的个体化根除治疗是十分必要的。
3、目前,可用于h.pylori个体化治疗药敏指导的检测方法主要包括:基于培养的表型药敏检测方法,例如琼脂如稀释法(金标准)、e-test法等以及基于耐药突变的分子药敏检测方法,例如pcr法等。然而,这些方法具有一定的局限性,其限制了药敏指导的个体化根除治疗的临床推广应用。
4、二代测序技术(nextgeneration sequencing,ngs)作为一种全面、具有成本效益、快速地获得细菌全基因组信息的工具,可以用于耐药性感染性疾病监测和进化分析,在临床分离株中发现罕见耐药机制具有潜力。近年来,随着测序成本的下降及生物信息学技术的发展,一种结合细菌ngs及机器学习预测耐药结局并识别耐药相关特征的方法被开发出来。这种方法无需依赖预先存在的amr基因或突变的数据库,通过泛基因组的方法将ngs全基因组序列与耐药表型信息关联,最终获得耐药预测模型,其可行性已在大肠杆菌、结核分枝杆菌、沙门氏菌以及铜绿假单胞菌等致病菌中得以实现。进一步利用shapley additiveexplanation(shap)values对机器学习的“黑箱模型”进行可视化,揭示影响机器学习模型的每个重要特征。然而,上述细菌耐药预测策略能否应用于h.pylori多药耐药预测,成为本领域技术人员亟需解决的问题。
技术实现思路
1、为此,本申请提供一种幽门螺杆菌对多药耐药表型的预测模型构建方法及装置,以解决现有技术存在的用于h.pylori个体化治疗药敏指导的检测方法限制了药敏指导的个体化根除治疗的临床推广应用的问题。
2、为了实现上述目的,本申请提供如下技术方案:
3、第一方面,一种幽门螺杆菌对多药耐药表型的预测模型构建方法,包括:
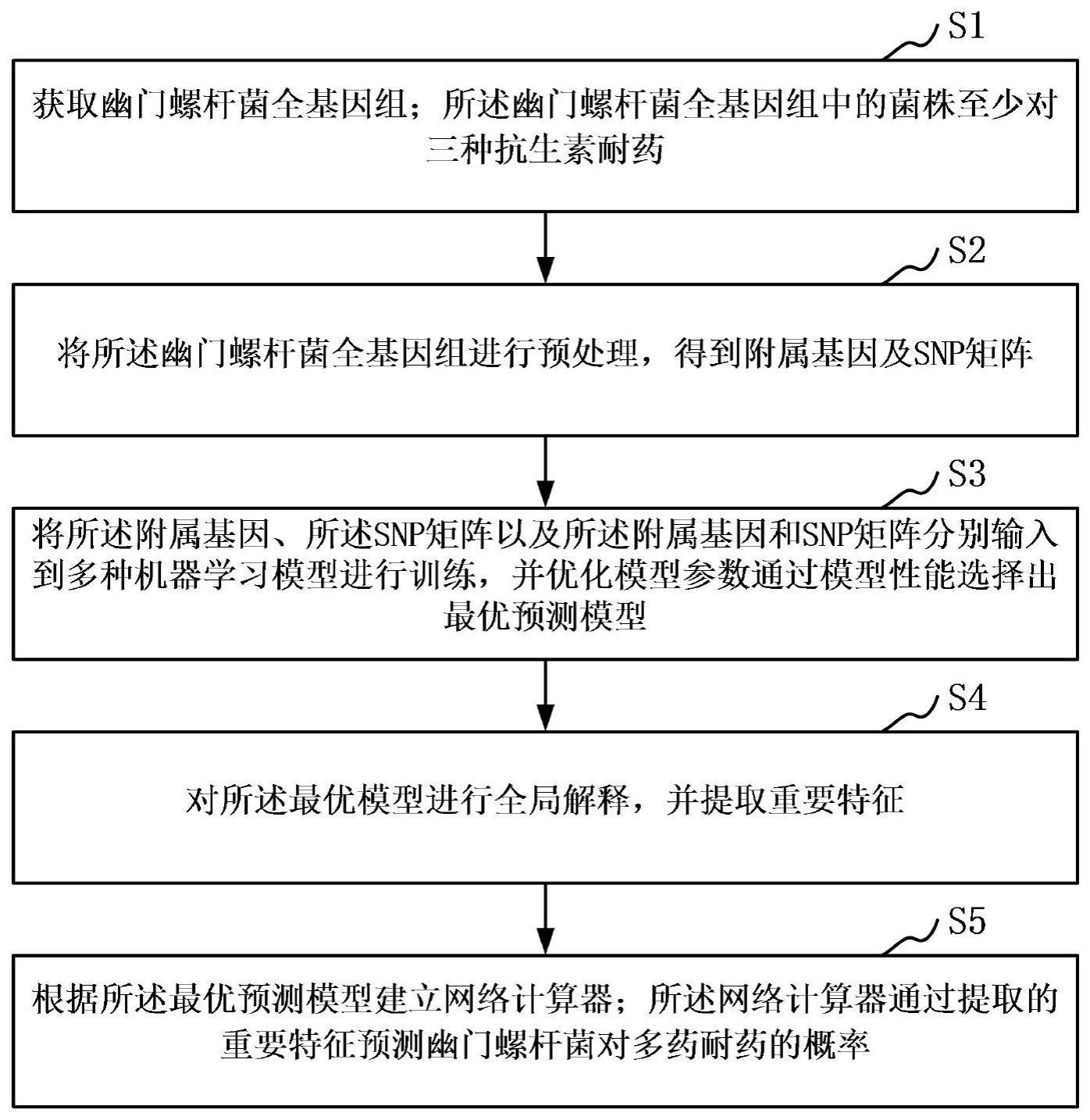
4、步骤1:获取幽门螺杆菌全基因组;所述幽门螺杆菌全基因组中的菌株至少对三种抗生素耐药;
5、步骤2:将所述幽门螺杆菌全基因组进行预处理,得到附属基因及snp矩阵;
6、步骤3:将所述附属基因、所述snp矩阵以及所述附属基因和snp矩阵分别输入到多种机器学习模型进行训练,并优化模型参数通过模型性能选择出最优预测模型;
7、步骤4:对所述最优模型进行全局解释,并提取重要特征;
8、步骤5:根据所述最优预测模型建立网络计算器;所述网络计算器通过提取的重要特征预测幽门螺杆菌对多药耐药的概率。
9、作为优选,所述步骤2包括:
10、步骤201:将所述幽门螺杆菌全基因组进行基因组注释,得到核心基因和附属基因;
11、步骤202:识别所述核心基因中的单核苷酸多态,得到snp矩阵。
12、作为优选,所述步骤201中,采用prokka将所述幽门螺杆菌全基因组进行基因组注释。
13、作为优选,所述步骤3中,多种机器学习模型包括knn、lr、svm、rf、gbdt以及xgboost。
14、作为优选,所述步骤3中,优化模型参数时采用网格搜索和交叉验证的方法进行优化。
15、作为优选,所述步骤4中,采用shap对所述最优预测模型进行全局解释。
16、作为优选,所述步骤4中,提取的重要特征包括rlpathr216lys、group_1364、glmuglu162thr、hp_0731asn511asp、smc、gspa、group_333、polaval112thr、omp13serleu10phephe和hp_0922ser2141ala。
17、第二方面,一种幽门螺杆菌对多药耐药表型的预测模型构建装置,包括:
18、数据获取模块,用于获取幽门螺杆菌全基因组;所述幽门螺杆菌全基因组中的菌株至少对三种抗生素耐药;
19、数据预处理模块,用于将所述幽门螺杆菌全基因组进行预处理,得到附属基因及snp矩阵;
20、训练模块,用于将所述附属基因、所述snp矩阵以及所述附属基因和snp矩阵分别输入到多种机器学习模型进行训练,并优化模型参数通过模型性能选择出最优预测模型;
21、全局解释模块,用于对所述最优模型进行全局解释,并提取重要特征;
22、预测模块,用于根据所述最优预测模型建立网络计算器;所述网络计算器通过提取的重要特征预测幽门螺杆菌对多药耐药的概率。
23、第三方面,一种计算机设备,包括存储器和处理器,所述存储器存储有计算机程序,所述处理器执行所述计算机程序时实现幽门螺杆菌对多药耐药表型的预测模型构建方法的步骤。
24、第四方面,一种计算机可读存储介质,其上存储有计算机程序,所述计算机程序被处理器执行时实现幽门螺杆菌对多药耐药表型的预测模型构建方法的步骤。
25、相比现有技术,本申请至少具有以下有益效果:
26、本申请提供了一种幽门螺杆菌对多药耐药表型的预测模型构建方法及装置,通过获取幽门螺杆菌全基因组,幽门螺杆菌全基因组中的菌株至少对三种抗生素耐药;将幽门螺杆菌全基因组进行预处理,得到附属基因及snp矩阵;将附属基因、snp矩阵以及附属基因和snp矩阵分别输入到多种机器学习模型进行训练,并优化模型参数通过模型性能选择出最优预测模型;对最优模型进行全局解释,并提取重要特征;根据最优预测模型建立网络计算器;网络计算器通过提取的重要特征预测幽门螺杆菌对多药耐药的概率。通过本申请构建的网络计算器能够预测幽门螺杆菌对多药耐药的概率,并用于指导h.pylori临床根除决策和个体化用药方案选择。
技术特征:
1.一种幽门螺杆菌对多药耐药表型的预测模型构建方法,其特征在于,包括:
2.根据权利要求1所述的幽门螺杆菌对多药耐药表型的预测模型构建方法,其特征在于,所述步骤2包括:
3.根据权利要求2所述的幽门螺杆菌对多药耐药表型的预测模型构建方法,其特征在于,所述步骤201中,采用prokka将所述幽门螺杆菌全基因组进行基因组注释。
4.根据权利要求1所述的幽门螺杆菌对多药耐药表型的预测模型构建方法,其特征在于,所述步骤3中,多种机器学习模型包括knn、lr、svm、rf、gbdt以及xgboost。
5.根据权利要求1所述的幽门螺杆菌对多药耐药表型的预测模型构建方法,其特征在于,所述步骤3中,优化模型参数时采用网格搜索和交叉验证的方法进行优化。
6.根据权利要求1所述的幽门螺杆菌对多药耐药表型的预测模型构建方法,其特征在于,所述步骤4中,采用shap对所述最优预测模型进行全局解释。
7.根据权利要求1所述的幽门螺杆菌对多药耐药表型的预测模型构建方法,其特征在于,所述步骤4中,提取的重要特征包括rlpathr216lys、group_1364、glmu glu162thr、hp_0731asn511asp、smc、gspa、group_333、polaval112thr、omp13 serleu10phephe和hp_0922ser2141ala。
8.一种幽门螺杆菌对多药耐药表型的预测模型构建装置,其特征在于,包括:
9.一种计算机设备,包括存储器和处理器,所述存储器存储有计算机程序,其特征在于,所述处理器执行所述计算机程序时实现权利要求1至7中任一项所述的方法的步骤。
10.一种计算机可读存储介质,其上存储有计算机程序,其特征在于,所述计算机程序被处理器执行时实现权利要求1至7中任一项所述的方法的步骤。
技术总结
本申请公开了一种幽门螺杆菌对多药耐药表型的预测模型构建方法及装置,涉及耐药性预测技术领域,通过将附属基因、SNP矩阵以及附属基因和SNP矩阵分别输入到多种机器学习模型进行训练,并优化模型参数通过模型性能选择出最优预测模型;对最优模型进行全局解释,并提取重要特征;根据最优预测模型建立网络计算器;网络计算器通过提取的重要特征预测幽门螺杆菌对多药耐药的概率。本申请利用H.pylori全基因组数据结合机器学习算法构建H.pylori多药耐药表型的预测模型,利用SHAP对预测模型进行可视化,并基于此构建网络计算器,能够用于指导H.pylori临床根除决策和个体化用药方案选择。
技术研发人员:袁媛,宫月华,王迎迎
受保护的技术使用者:中国医科大学附属第一医院
技术研发日:
技术公布日:2024/1/15
- 还没有人留言评论。精彩留言会获得点赞!