本发明涉及生物,具体涉及一种基于rna序列和结构信息特征预测rna与小分子结合点位模型的构建方法及其应用。
背景技术:
1、近年来rna小分子作为治疗疾病的潜在靶点逐渐受到关注,rna-小分子结合位点对靶向药物的研究至关重要,然而由于rna的序列和结构的复杂性较高,预测rna-小分子结合点位仍然具有巨大的挑战。
2、因为现有的方法对真正样本的识别度不是很高,所以我们基于rna的序列和结构的特征提出了一个新的预测方法:rnabind。在两个独立的测试集上的实验结果表明rnabind整体的性能优于现有的方法,特别是极大地提高了recall,使模型能够更好地捕捉到真正样本。rnabind有望成为设计rna靶向小分子药物的有用工具。
3、rna分子的功能复杂多样,对维持生命活动和机体功能起着至关重要的作用。rna小分子可以通过与rna大分子的特定结合点位相互作用,发挥调控基因表达、干扰rna结构等功能。在药物研发领域,靶向rna的小分子药物(rna-targeted small molecules)具有针对rna结构或功能的特异性结合能力,可用于调控rna的生物学活性,有望作为治疗相关疾病的新型药物,这促进了rna与小分子结合点位预测的计算方法的发展。
4、由于rna的序列和结构的多样性,加上rna-小分子结合点位数据较为稀少,导致只有少数的几种方法被开发出来,包括rsite,rsite2,rbind,rnasite和rlbind。rsite是一种基于rna三级结构的计算方法,它首先计算每个核苷酸和rna分子中所有其他核苷酸之间的欧氏距离,然后确定距离曲线中的极值点作为功能位点的核苷酸。rsite2则是在rsite的基础上使用rna的二级结构来进行预测。rbind是一种基于结构的计算网络,使用度和结合贴近度来识别rna-小分子的结合点位。rnasite和rlbind则都使用了rna的序列和结构特征来预测rna-小分子的结合点位。就目前最新的两种方法来说,虽然他们都使用了rna的序列和结构的特征,但是对真正样本的识别度不是很高。
技术实现思路
1、本发明所要解决的技术问题在于如何提供一种预测rna与小分子结合点位模型的构建方法。
2、本发明通过以下技术手段实现解决上述技术问题的:
3、本发明的第一方面提出一种基于rna序列和结构信息特征预测rna与小分子结合点位模型的构建方法,包括以下步骤:
4、s1、建模数据收集:从pdb数据库中收集rna与小分子相互作用rna复合物的id,经过处理后作为建模样本;
5、s2、数据集的划分:将所有rna-小分子相互作用样本按照比例分为训练集和测试集;
6、s3、数据正负集的划分:核苷酸和小分子之间的原子距离小于特定的距离截止值则核苷酸被定义为与小分子相互作用,即正样本,其余为负样本;
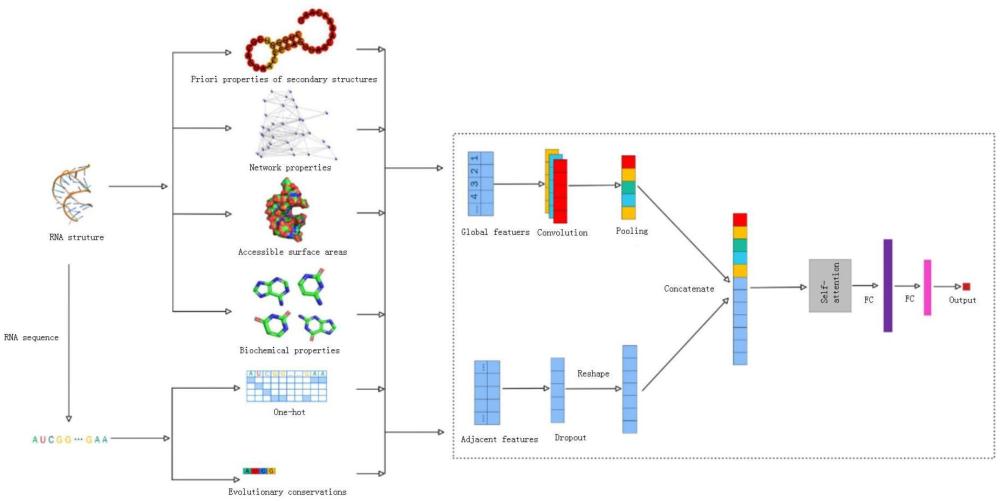
7、s4、rna特征的组合:选择rna序列的one-hot编码和序列中每个核苷酸的进化保守性评分作为序列特征;选择点位先验性质、网络拓扑性质、生化性质和asa编码作为结构特征;将特征组合后获得具有rna序列和结构信息的综合特征;
8、s5、模型构建:使用卷积模块、滑动窗口模块和自注意力模块组成的深度学习模型,对具有rna序列和结构特征的综合特征进行预测并得出结果。
9、优选的,所述s1中,处理的方法为如果rna分子与一个或多个配体形成复合物,则选择复合物进行结合位点预测。
10、优选的,所述s1中,处理的方法为使用结构相似性聚类去除冗余结构,成对结构相似性tm-scorerna的截断值为0.3。
11、优选的,所述s1中,处理的方法为模拟rna三级结构,通过过滤具有多链或假结相互作用的rna结构来保留rna。
12、优选的,所述s5中,采用卷积模块来提取rna编码的全局特征;采用滑动窗口模块来提取rna编码的局部特征;最后将全局与局部特征拼接输入到自注意力模块得出预测结果。
13、本发明的第二方面提出一种基于rna序列和结构信息的小分子结合点位的预测方法,该预测方法采用上述方法构建的模型来实现。
14、优选的,包括以下步骤:
15、(1)分别对rna的每个核苷酸序列与结构特征进行计算编码;
16、(2)将rna的每个核苷酸序列与结构特征进行拼接,输入到所述小分子结合点位预测模型中,得出rna每个核苷酸为小分子结合点位的概率值;
17、(3)根据阈值确定核苷酸是否为小分子结合点位,大于阈值判定为结合点位。
18、优选的,所述步骤(1)中,序列特征包括对序列one-hot编码,使用consurfdb算法来确定每一个核苷酸的进化保守性评分;结构特征包括使用二级结构相似性点位先验算法计算结合点位在rna二级结构中的概率、使用三级结构的网络拓扑算法计算三级结构中每个节点的重要性、使用rnasol算法来计算核苷酸的asa。
19、优选的,所述步骤(2)中,每个rna核苷酸的编码长度为16d,每条rna固定长度为64,输入到模型得到每个rna包含64×16个特征信息。
20、优选的,所述步骤(3)中,将每条rna64×16个特征信息输入到模型中,得出模型对每一个核苷酸是否为小分子结合点位。
21、本发明的有益效果在于:
22、本发明提出一种全新的预测方法rnabind来预测rna-小分子结合点位。在这项工作中我们使用rna的序列、二级结构、三级结构和生化性质,序列的特征包括了对核苷酸类型的one-hot编码和对核苷酸进行进化保守性评分。结构的特征包括了二级结构相似性的点位先验性质、三级结构的网络拓扑性质和可接触表面积(asa),其中二级结构是我们设计的编码方式来对二级结构相似的rna进行特征提取。最后生化性质包括了对rna分子量和侧链pka的编码。极大提高了预测rna-小分子的结合点位的效率和准确性。在两个独立的测试集上的实验结果表明rnabind整体的性能优于现有的方法,特别是极大地提高了recall,使模型能够更好地捕捉到真正样本。rnabind有望成为设计rna靶向小分子药物的有用工具。
技术特征:1.一种基于rna序列和结构信息特征预测rna与小分子结合点位模型的构建方法,其特征在于,包括以下步骤:
2.根据权利要求1所述的构建方法,其特征在于,所述s1中,处理的方法为如果rna分子与一个或多个配体形成复合物,则选择复合物进行结合位点预测。
3.根据权利要求1所述的构建方法,其特征在于,所述s1中,处理的方法为使用结构相似性聚类去除冗余结构,成对结构相似性tm-scorerna的截断值为0.3。
4.根据权利要求1所述的构建方法,其特征在于,所述s1中,处理的方法为模拟rna三级结构,通过过滤具有多链或假结相互作用的rna结构来保留rna。
5.根据权利要求1所述的构建方法,其特征在于,所述s5中,采用卷积模块来提取rna编码的全局特征;采用滑动窗口模块来提取rna编码的局部特征;最后将全局与局部特征拼接输入到自注意力模块得出预测结果。
6.一种基于rna序列和结构信息的小分子结合点位的预测方法,其特征在于,该预测方法采用权利要求1-5任一项所述的方法构建的模型来实现。
7.根据权利要求6所述的预测方法,其特征在于,包括以下步骤:
8.根据权利要求1所述的预测方法,其特征在于,所述步骤(1)中,序列特征包括对序列one-hot编码,使用consurfdb算法来确定每一个核苷酸的进化保守性评分;结构特征包括使用二级结构相似性点位先验算法计算结合点位在rna二级结构中的概率、使用三级结构的网络拓扑算法计算三级结构中每个节点的重要性、使用rnasol算法来计算核苷酸的asa。
9.根据权利要求6所述的预测方法,其特征在于,所述步骤(2)中,每个rna核苷酸的编码长度为16d,每条rna固定长度为64,输入到模型得到每个rna包含64×16个特征信息。
10.根据权利要求6所述的预测方法,其特征在于,所述步骤(3)中,将每条rna 64×16个特征信息输入到模型中,得出模型对每一个核苷酸是否为小分子结合点位。
技术总结本发明公开了一种基于RNA序列和结构信息特征预测RNA与小分子结合点位模型的构建方法及其应用,属于生物技术领域。该模型的构建方法包括以下步骤:S1、建模数据收集;S2、数据集的划分;S3、数据正负集的划分;S4、RNA特征的组合;S5、模型构建。还提出一种基于RNA序列和结构信息的小分子结合点位的预测方法,该预测方法采用上述方法构建的模型来实现。有益效果:极大提高了预测RNA‑小分子的结合点位的效率和准确性。在两个独立的测试集上的实验结果表明RNABind整体的性能优于现有的方法,特别是极大地提高了recall,使模型能够更好地捕捉到真正样本。RNABind有望成为设计RNA靶向小分子药物的有用工具。
技术研发人员:孙战里,丁文杰
受保护的技术使用者:安徽大学
技术研发日:技术公布日:2025/1/9