基于图transformer的单细胞RNA测序数据聚类方法
本发明涉及深度学习和生物信息技术,具体涉及基于图transformer的单细胞rna测序数据聚类方法。
背景技术:
1、作为当前最热门的生命组学技术之一,单细胞rna测序(scrna-seq)能够在单细胞分辨率下研究细胞状态、通讯以及异质性。scrna-seq分析的首要步骤是利用细胞聚类鉴定生物系统包含的细胞类型。随着深度学习算法的发展,图神经网络(gnn)已经被广泛用于scrna-seq数据分析,以提取每个细胞的特征向量并进行细胞聚类,凭借其强大的图表示学习能力,已经成为生物学网络和单细胞数据分析领域的重要方法。
2、当前已经出现了多种推断scrna-seq数据细胞特异性网络的算法,如csn、c-csn、loccsn等,这些算法的第一步都是构建一个细胞网络(或包含细胞的异构网络),再利用gnn学习网络中每个细胞的节点嵌入,这种方法更善于利用细胞间的关联性,简单的连接关系无法充分反映细胞间的真实差异,这会导致gnn在更新节点表示时,无法准确地捕捉和表示这些异质性信息,难以区分基因间的相关性属于直接关联还是间接关联,也无法识别相关性源于哪种具体的分子相互作用,这导致现有的网络包含大量被高估的间接关联不容易捕获不同细胞间潜在的异质性,导致模型在预测细胞类型、功能或状态等方面的准确性下降,并会限制模型应用范围。
技术实现思路
1、本发明的目的是提供基于图transformer的单细胞rna测序数据聚类方法,以解决现有技术中的上述不足之处。
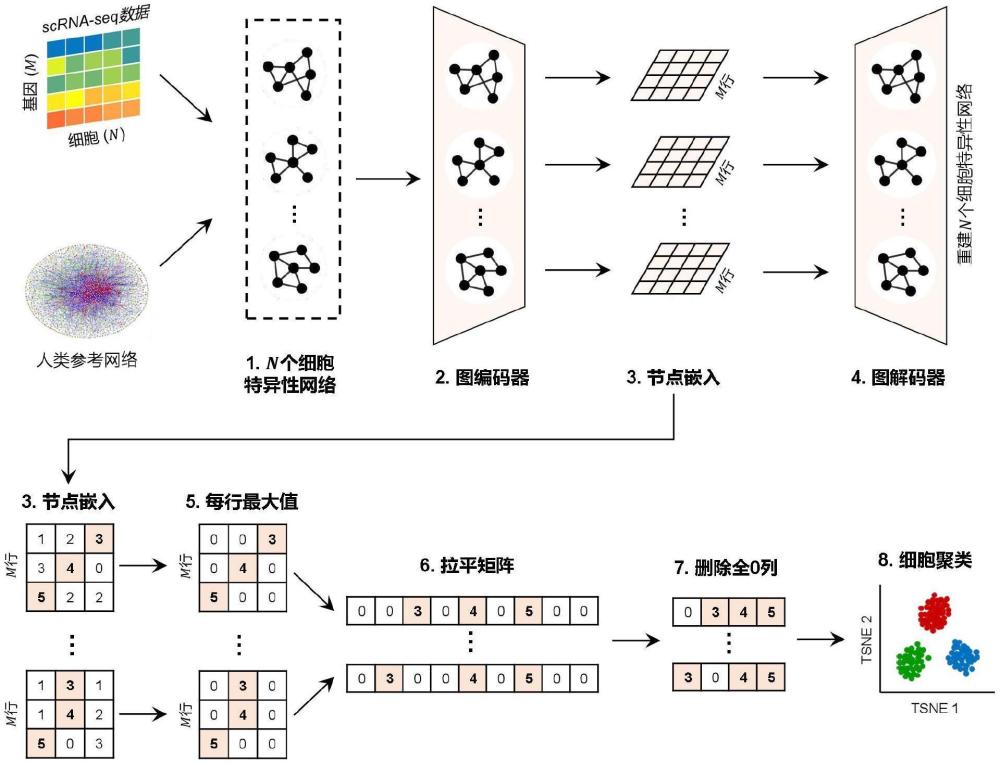
2、为了实现上述目的,本发明提供如下技术方案:基于图transformer的单细胞rna测序数据聚类方法,包括如下步骤:
3、s1、首先根据scrna-seq数据构建细胞特异性信号网络;
4、s2、然后用细胞特异性信号网络训练同一个图神经网络,以学习细胞特有的图表示,从而得到细胞特异性信号网络的节点嵌入矩阵;
5、s3、最后根据节点嵌入矩阵获得细胞特异性网络的图嵌入向量,基于图嵌入进行基于图表示的细胞聚类,得到聚类结果。
6、进一步地,s1所述首先根据scrna-seq数据构建细胞特异性信号网络,包括如下步骤:
7、s11、构建人类分子相互作用参考网络:从24个数据库中收集4种已知的人类分子相互作用信息,对收集到的相互作用信息保留至少2个数据库共享的高可信预测,并基于上述相互作用建立一个集成了多维分子相互作用的参考网络;
8、s12、数据预处理:通过python的scanpy包对scrna-seq数据进行预处理,得到每个细胞的基因和基因表达量;
9、s13、筛选每个细胞的激活基因:将每个细胞的基因按表达量从高到低进行排序,筛选表达量前5%的基因为每个细胞的激活基因;
10、s14、从参考网络中提取细胞特异性子网,构成细胞特异性的信号网络:针对每个细胞的激活基因,从s11所述参考网络中提取关联激活基因的相互作用,组成细胞特异性信号网络。
11、进一步地,s11所述相互作用信息包括配体-受体相互作用、蛋白质-蛋白质相互作用、转录因子-靶基因相互作用和生物学通路。
12、进一步地,s12所述预处理包括过滤缺失值超过99%的基因和细胞,归一化不同细胞的基因表达并对基因表达进行对数转化。
13、进一步地,s2所述然后用细胞特异性信号网络训练同一个图神经网络,以学习细胞特有的图表示,从而得到细胞特异性信号网络的节点嵌入矩阵,包括如下步骤:
14、s21、构建基于图transformer的图自编码器:所述编码器接收单细胞网络的邻接矩阵和节点的特征矩阵两类输入;所述编码器包含两层,第1层的输入维度为细胞特异性信号网络中节点的特征向量的维度,输出维度为32维,采用relu激活函数;第2层的输入和输出维度均为32维,采用sigmoid激活函数,所述编码器能够捕捉细胞特异性信号网络中的局部和全局拓扑结构,从而为每个节点生成32维的节点嵌入;
15、s22、解码器构建:通过计算节点嵌入之间的内积,来预测和重建细胞特异性信号网络中的连接关系,所述解码器能够重现输入的细胞特异性信号网络,使得输出网络的拓扑结构与输入细胞特异性信号网络一致;
16、s23、损失函数优化:在学习过程中通过损失函数来优化模型参数,损失函数衡量的是解码器重建的网络与输入细胞特异性信号网络之间的差异,并将该差异最小化,得到每个细胞网络的节点嵌入矩阵。
17、进一步地,s22所述预测和重建基因网络中的连接关系,采用的是基于pytorch的innerproductdecoder函数。
18、进一步地,s3所述最后根据节点嵌入矩阵获得细胞特异性网络的图嵌入向量,基于图嵌入进行基于图表示的细胞聚类,得到聚类结果,包括如下步骤:
19、s31、基于每个细胞网络节点嵌入矩阵,将每行最大元素之外的其它元素都变为0,然后将2维矩阵拉平为1维向量,作为每个细胞的图嵌入向量。
20、s32、然后整合所有细胞的图嵌入向量构成图嵌入矩阵,将图嵌入矩阵预处理后进行聚类。
21、进一步地,s32所述图嵌入矩阵包含每个细胞的图表示特征。
22、进一步地,s32所述预处理为首先删除矩阵中全0的列,然后使用主成分分析方法将图嵌入矩阵进行降维。
23、进一步地,s32所述聚类采用的是louvain聚类方法或k-means聚类方法。
24、与现有技术相比,本发明提供的基于图transformer的单细胞rna测序数据聚类方法,通过构建每个细胞的特异性网络,再利用gnn学习每个细胞的特异性图嵌入,以揭示细胞间的异质性,扩大模型的应用范围;
25、根据每个细胞的独特基因表达模式和调控特征来为每个细胞构建一个特异性的信号网络,每个细胞的特异性网络都反映了其独特的生物学状态和潜在的功能特性;
26、利用gnn的强大学习能力,捕捉图结构中的复杂信息,包括细胞之间的连接和属性,每个细胞都可以被映射到一个高维空间中的特定位置,这个位置(即细胞的特异性图嵌入)准确地反映了其生物学特性和与其他细胞的异质性;
27、这种新的细胞聚类方法不仅提高了聚类的准确性和鲁棒性,还为后续的生物学分析和研究提供了丰富的信息。例如通过比较不同细胞的特异性图嵌入,可以揭示出潜在的细胞亚群或新的细胞类型;通过分析细胞间的嵌入距离,可以推断出它们之间的相互作用和调控关系。
技术特征:
1.基于图transformer的单细胞rna测序数据聚类方法,其特征在于,包括如下步骤:
2.根据权利要求1所述的基于图transformer的单细胞rna测序数据聚类方法,其特征在于,s1所述首先根据scrna-seq数据构建细胞特异性信号网络,包括如下步骤:
3.根据权利要求2所述的基于图transformer的单细胞rna测序数据聚类方法,其特征在于,s11所述相互作用信息包括配体-受体相互作用、蛋白质-蛋白质相互作用、转录因子-靶基因相互作用和生物学信号通路。
4.根据权利要求2所述的基于图transformer的单细胞rna测序数据聚类方法,其特征在于,s12所述预处理包括过滤缺失值超过99%的基因和细胞,归一化不同细胞的基因表达并对基因表达进行对数转化。
5.根据权利要求1所述的基于图transformer的单细胞rna测序数据聚类方法,其特征在于,s2所述然后用细胞特异性信号网络训练同一个图神经网络,以学习细胞特有的图表示,从而得到细胞特异性信号网络的节点嵌入矩阵,包括如下步骤:
6.根据权利要求5所述的基于图transformer的单细胞rna测序数据聚类方法,其特征在于,s22所述预测和重建基因网络中的连接关系,采用的是基于pytorch的innerproductdecoder函数。
7.根据权利要求1所述的基于图transformer的单细胞rna测序数据聚类方法,其特征在于,s3所述最后根据节点嵌入矩阵获得细胞特异性网络的图嵌入向量,基于图嵌入进行基于图表示的细胞聚类,得到聚类结果,包括如下步骤:
8.根据权利要求7所述的基于图transformer的单细胞rna测序数据聚类方法,其特征在于,s32所述图嵌入矩阵包含每个细胞的图表示特征。
9.根据权利要求7所述的基于图transformer的单细胞rna测序数据聚类方法,其特征在于,s32所述预处理步骤包括:首先删除矩阵中全0的列,然后使用主成分分析方法将图嵌入矩阵进行降维。
10.根据权利要求7所述的基于图transformer的单细胞rna测序数据聚类方法,其特征在于,s32所述聚类所采用的方法为louva i n聚类方法或k-means聚类方法。
技术总结
本发明公开了基于图transformer的单细胞RNA测序数据聚类方法,涉及深度学习和生物信息技术领域,包括如下步骤:首先根据scRNA‑seq数据构建细胞特异性信号网络;然后用细胞特异性信号网络训练同一个图神经网络,以学习细胞特有的图表示,从而得到细胞特异性信号网络的节点嵌入矩阵;最后根据节点嵌入矩阵进行基于图表示的细胞聚类;该基于图transformer的单细胞RNA测序数据聚类方法,通过构建每个细胞的特异性网络,再利用GNN学习每个细胞的特异性图嵌入,以揭示细胞间的异质性,扩大模型的应用范围;这种新的细胞聚类方法不仅提高了聚类的准确性和鲁棒性,还为后续的生物学分析和研究提供丰富的信息。
技术研发人员:朱云平,韩明飞,李晓,陈涛,赵羽
受保护的技术使用者:中国人民解放军军事科学院军事医学研究院
技术研发日:
技术公布日:2025/3/16
- 还没有人留言评论。精彩留言会获得点赞!