一种2,4,6-三甲基苯甲酸的制备方法与流程
本发明涉及化工产品的制备方法,尤其涉及一种2,4,6-三甲基苯甲酸的制备方法。
背景技术:
2,4,6三甲基苯甲酸是一种重要的有机合成中间体,广泛应用于生产染料、杀虫剂、医药和光引发剂的中间体。近年来,随着其应用领域不断增加,2,4,6三甲基苯甲酸的市场潜力也越来越大,工业化制备越来越受到人们的广泛关注。
目前合成2,4,6-三甲基苯甲酸的方法主要为均三甲苯二氧化碳直接羧化法,即以无水氯化铝为催化剂,催化均三甲苯和二氧化碳合成2,4,6-三甲基苯甲酸的方法。但是由上述反应过程中,需要使用大量的无水氯化铝,而且制备得到的2,4,6-三甲基苯甲酸纯度较低,约为92-96%,无法满足人们需求。
技术实现要素:
本发明提供了一种2,4,6-三甲基苯甲酸的制备方法,本发明提供的制备方法以氯化氢或浓盐酸为催化剂,避免了现有技术中以无水氯化铝为催化剂时,需要使用大量无水氯化铝的问题,而且本发明提供的方法得到的2,4,6-三甲基苯甲酸纯度较高,可达98.1%~99.1%。
本申请提供了一种2,4,6-三甲基苯甲酸的制备方法,包括以下步骤:
将均三甲苯和二氧化碳在催化剂作用下进行反应,得到2,4,6-三甲基苯甲酸;所述催化剂为氯化氢或浓盐酸。
优选的,所述浓盐酸的质量浓度为37%~40%。
优选的,所述催化剂的质量为均三甲苯质量的0.1%~20%。
优选的,所述均三甲苯和二氧化碳的摩尔比为1:0.3~0.5。
优选的,采用向反应器连续注入均三甲苯和二氧化碳的方式进行反应;所述均三甲苯的连续注入流量为50~100ml/min。
优选的,所述反应的温度为20~70℃;所述反应的压力为2~4mpa。
优选的,所述反应的时间为5~25s。
优选的,所述反应后还包括对反应产物的后处理;
当所述催化剂为氯化氢时,所述后处理为:将所述反应产物进行冷却和固液分离,得到2,4,6-三甲基苯甲酸;
当所述催化剂为浓盐酸时,所述后处理为:将所述反应产物依次进行分层、有机层冷却和固液分离,得到2,4,6-三甲基苯甲酸。
本发明提供了一种2,4,6-三甲基苯甲酸的制备方法,包括以下步骤:将均三甲苯和二氧化碳在催化剂作用下进行反应,得到2,4,6-三甲基苯甲酸;所述催化剂为氯化氢或浓盐酸。本发明提供的制备方法中,均三甲苯和二氧化碳在氯化氢或浓盐酸催化剂作用下发生反应,生成2,4,6-三甲基苯甲酸。本发明提供的制备方法采用氯化氢或浓盐酸代替现有技术中的无水氯化铝,避免了反应过程中需要使用大量无水氯化铝的问题,而且本发明提供的方法制备得到的2,4,6-三甲基苯甲酸纯度较高,可达98.1%~99.1%。另外,本发明提供的制备方法可连续反应,适合工业大规模生产。
附图说明
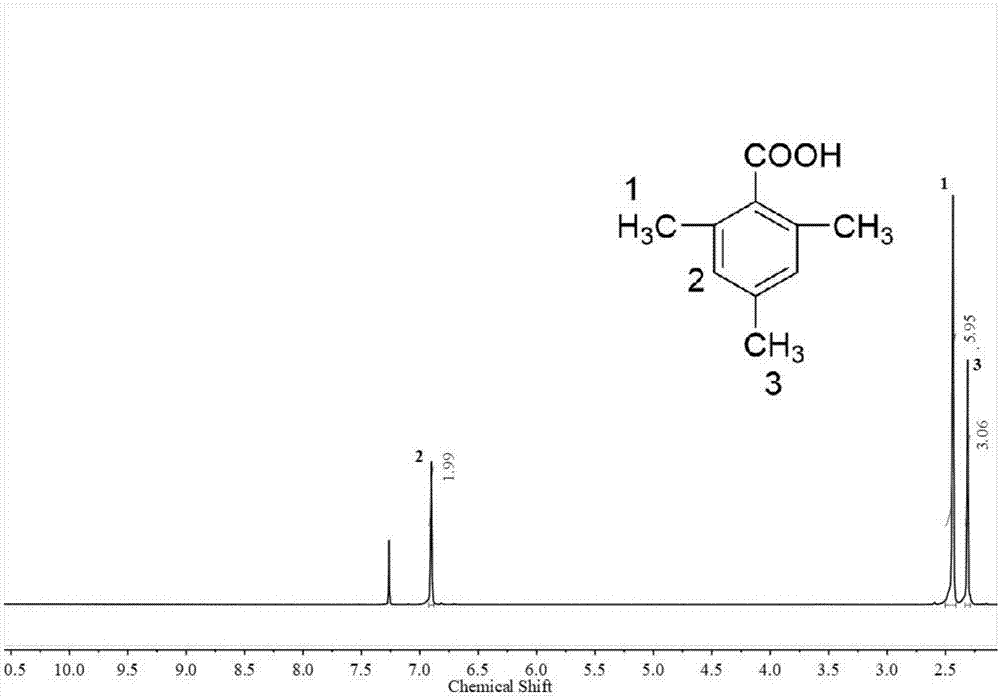
图1为本发明实施例1制备得到的2,4,6-三甲基苯甲酸的核磁谱图;
图2为本发明实施例1制备得到的2,4,6-三甲基苯甲酸的质谱图。
具体实施方式
本发明提供了一种2,4,6-三甲基苯甲酸的制备方法,包括以下步骤:
将均三甲苯和二氧化碳在催化剂作用下进行反应,得到2,4,6-三甲基苯甲酸;所述催化剂为氯化氢或浓盐酸。
在本发明中,如无特殊说明,所有原料均为市售商品。
本发明将均三甲苯和二氧化碳在催化剂作用下进行反应,得到2,4,6-三甲基苯甲酸。
在本发明中,所述二氧化碳优选为液态二氧化碳。在本发明中,所述催化剂为氯化氢或浓盐酸;当所述催化剂为浓盐酸时,所述浓盐酸的质量浓度优选为37%~40%,进一步优选为38%~39%。所述催化剂的质量为均三甲苯质量的0.1%~20%,进一步优选为5%~10%。
在本发明中,所述反应的压力优选为2~4mpa,进一步优选为2.5~3.5mpa,更优选为3.0~3.5mpa。本发明优选通过均三甲苯和二氧化碳的流量来控制反应的压力。本发明优选将反应压力控制在上述范围内,有利于反应顺利进行,生成2,4,6-三甲基苯甲酸。
在本发明中,所述均三甲苯和二氧化碳的摩尔比优选为1:0.3~0.5,进一步优选为1:0.35~0.45。本发明优选采用连续注入均三甲苯和二氧化碳的方式进行反应,使得反应能够连续进行;所述均三甲苯的连续注入流量优选为50~100ml/min,进一步优选为60~90ml/min,更优选为70~80ml/min。本发明优选将均三甲苯的连续注入流量控制在上述范围内,有利于将反应的压力控制在合适范围,有利于反应生成2,4,6-三甲基苯甲酸。
在本发明中,所述均三甲苯和二氧化碳优选同时连续注入反应器。本发明优选采用上述方式进行反应,有利于使本发明提供的制备方法可连续反应,适合工业大规模生产。
在本发明中,所述反应的温度优选为20~70℃,进一步优选为30~60℃,更优选为40~50℃。本发明优选将反应温度控制在上述范围内,一方面有利于使均三甲苯和二氧化碳反应生成2,4,6-三甲基苯甲酸;另一方面有利于使反应生成的产物2,4,6-三甲基苯甲酸完全溶解于均三甲苯中,便于将反应产物从反应装置中输送出去。
在本发明中,所述反应的时间优选为5~25s,进一步优选为10~20s,更优选为15~20s。在本发明中,当采用连续注入均三甲苯和二氧化碳的方式进行反应时,所述反应的时间以均三甲苯和二氧化碳注入开始计;本发明优选通过反应装置中盘管长度控制反应的时间。在本发明中,所述反应装置优选为微反应器;本发明对所述微反应器的来源没有特殊要求,采用本领域技术人员所熟知的即可。
在本发明中,所述反应优选用式i表示:
完成反应后,本发明优选将反应产物进行后处理,得到2,4,6-三甲基苯甲酸。
当所述催化剂为氯化氢时,本发明优选将所述反应产物进行冷却和固液分离,得到2,4,6-三甲基苯甲酸。在本发明中,所述冷却的温度优选为0~5℃。本发明优选通过冷却处理,使2,4,6-三甲基苯甲酸从反应体系中析出。本发明对冷却的具体实施方式没有特别限制,采用本领域技术人员所熟知的冷却方式即可。本发明优选将冷却后的反应产物进行固液分离,收集固体,得到2,4,6-三甲基苯甲酸。在本发明中,所述固液分离优选包括依次进行的离心和过滤。本发明对离心和过滤的具体实施方式没有特别限制,采用本领域技术人员所熟知的离心和过滤方式即可。在本发明中,所述过滤得到的滤液优选为均三甲苯,本发明优选将过滤得到的滤液输送回反应装置,进行回收利用,收集过滤得到的滤饼,即为2,4,6-三甲基苯甲酸。
当所述催化剂为浓盐酸时,本发明优选将所述反应产物依次进行分层、有机层冷却和固液分离处理,得到2,4,6-三甲基苯甲酸。在本发明中,所述反应产物会自动分成两层,上层为有机层,下层为盐酸层。本发明优选将盐酸层重新进行回收利用。
分层后,本发明优选将有机层依次进行有机层冷却和固液分离处理,得到2,4,6-三甲基苯甲酸。在本发明中,所述有机层冷却的温度优选为0~5℃。本发明优选通过冷却处理,使2,4,6-三甲基苯甲酸从有机层中析出,得到混合物。本发明对冷却的具体实施方式没有特别限制,采用本领域技术人员所熟知的冷却方式即可。本发明优选将冷却得到的混合物进行固液分离,收集固体,得到2,4,6-三甲基苯甲酸。在本发明中,所述固液分离优选包括依次进行的离心和过滤。本发明对离心和过滤的具体实施方式没有特别限制,采用本领域技术人员所熟知的离心和过滤方式即可。在本发明中,所述过滤得到的滤液优选为均三甲苯,本发明优选将过滤得到的滤液输送回反应装置,进行回收利用,收集过滤得到的滤饼,即为2,4,6-三甲基苯甲酸。
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。
实施例1
物料罐内加入各反应原料,控制均三甲苯流量为50ml/min,微反应器内温度为70℃,压力为3.0mpa,均三甲苯与二氧化碳摩尔比1:0.5,催化剂为质量浓度38%的浓盐酸,利用盘管长度控制反应时间为25s,反应结束后分层,下层浓盐酸回收,上层经冷却离心过滤,收集固体得到2,4,6-三甲基苯甲酸,过滤后的滤液输送回物料罐继续反应,产品纯度99.1%。
对实施例1制备得到的2,4,6-三甲基苯甲酸进行核磁分析和质谱分析,分析结果如图1和图2所示,其中图1为2,4,6-三甲基苯甲酸的核磁谱图,图2为2,4,6-三甲基苯甲酸的质谱图。由图1和图2可知,本发明制备得到的产物为2,4,6-三甲基苯甲酸。
实施例2
物料罐内加入各反应原料,控制均三甲苯流量为100ml/min,微反应器内温度为40℃,压力为3.5mpa,均三甲苯与二氧化碳摩尔比1:0.3,催化剂为氯化氢气体,利用盘管长度控制反应时间为25s,反应结束后经冷却离心过滤,收集固体得到2,4,6-三甲基苯甲酸,过滤后的滤液输送回物料罐继续反应,产品纯度99.0%。
实施例3
物料罐内加入各反应原料,控制均三甲苯流量为100ml/min,微反应器内温度为70℃,压力为2.0mpa,均三甲苯与二氧化碳摩尔比1:0.3,催化剂为质量浓度40%的浓盐酸,利用盘管长度控制反应的停留时间为20s,反应结束后分层,下层浓盐酸回收,上层经冷却离心过滤,收集固体得到2,4,6-三甲基苯甲酸,过滤后的滤液输送回物料罐继续反应,产品纯度98.1%。
实施例2和实施例3制备得到的2,4,6-三甲基苯甲酸的核磁谱图与质谱图与实施例1测试得到的图1和图2一致,在此不再赘述。
综上,本发明提供的方法能够制备得到纯度高的2,4,6-三甲基苯甲酸,纯度可达98.1~99.1%。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!