一种替西罗莫司的分离纯化方法与流程
本发明属于仿制药分离纯化领域,具体为一种替西罗莫司的分离纯化方法。
背景技术:
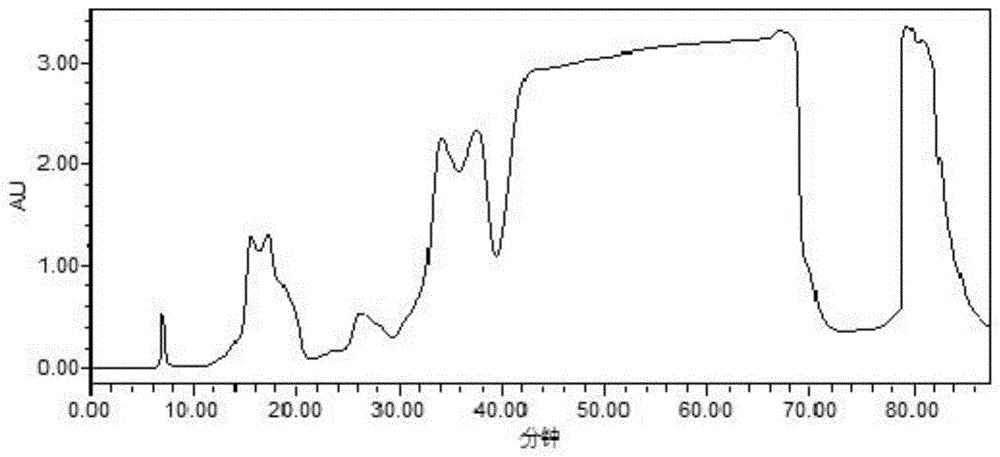
:替西罗莫司(坦西莫司)(temsirolimus)是一种静脉注射的mtor抑制剂,获批用来治疗晚期肾细胞癌,是西罗莫司的衍生物和前药。替西罗莫司以雷帕霉素(西罗莫司)为原料,通过酯化,脱保护等合成工艺生成替西罗莫司成品纯度低,异构体含量较高。目前研究报道的多为雷帕霉素(西罗莫司)的分离纯化方法(cn104844620a、cn107619413a等)。经reaxys检索发现替西罗莫司的分离纯化报道较少。郑从燊等在“第十三届全国抗生素学术会议(196页-201页)”公布了一种替西罗莫司的精制研究:将由半合成获得的替西罗莫司粗品经硅胶柱纯化、反相hplc纯化,再采用乙醚溶清,20℃下搅拌析晶,对替西罗莫司粗品进行精制,收率70%,替西罗莫司成品中异构体比例<0.5%。从报道的文献可以看出,目前对替西罗莫司分离纯化的研究较少,采用重结晶的方式能使异构体含量达到1.0%以下,但是乙醚沸点低且易燃易爆,不宜用于工业化大生产。技术实现要素:为了解决上述技术问题,本发明的目的在于针对现有合成工艺,提供一种替西罗莫司的分离纯化方法,它包括以下步骤:(1)粗品的合成:a.酰氯的制备:将2,2,5-三甲基-5-羧酸-1,3-二氧六烷溶解于正己烷,25±5℃下加入二氯亚砜,搅拌,减压浓缩至干,得酰氯;b.成酯反应:将雷帕霉素、dipea与dcm混合,0-5℃下滴入步骤a所得酰氯,25±5℃下搅拌,浓缩蒸干,粗品用硅胶柱层析,洗脱得酯化物2,2,5-三甲基[1,3-二氧六烷]-5-羧酸-42-酯-雷帕霉素;c.将步骤b酯化物溶解于thf,25±5℃下滴入硫酸水溶液,反应,调节ph至7-8,ea萃取,用干燥剂干燥,减压浓缩,得替西罗莫司粗品。(2)分离纯化:将步骤c粗品溶解于乙醇-正庚烷,注入正向制备色谱柱,用流动相乙醇-正庚烷梯度洗脱,分段收集洗脱液,合并替西罗莫司纯度≥98%的洗脱液,减压干燥,得替西罗莫司纯品;所述梯度洗脱程序为:0~25min,100%→50%正庚烷;25~70min,50%→50%正庚烷;70-90min,50%→20%正庚烷。进一步地,步骤a所述2,2,5-三甲基-5-羧酸-1,3-二氧六烷、正己烷与二氯亚砜的质量体积比为1g:14ml:1.37g;所述搅拌时间为12~15h;所述酰氯为无色油状液体。进一步地,步骤b所述雷帕霉素、dipea和dcm的质量体积比为1g:0.72ml:2.83ml;所述滴入酰氯至酰氯浓度为0.75~1g/ml;;所述搅拌至雷帕霉素含量小于0.5%时止;所述硅胶柱中硅胶的目数为200-300目;所述层析使用的洗脱剂是石油醚:乙酸乙酯。更进一步地,所述石油醚:乙酸乙酯体积比为1:1。进一步地,步骤c所述酯化物、thf与硫酸水溶液的质量体积比1g:7.95ml:1.88ml;所述硫酸水溶液浓度为2mol/l;所述反应时间为14h;所述调ph的试剂是饱和碳酸钠水溶液;所述干燥剂为无水硫酸钠。进一步地,步骤(2)所述粗品与乙醇-正庚烷的质量体积比为0.2:1g/ml;所述乙醇-正庚烷体积比:0~8:1~2。进一步地,步骤(2)所述正向制备色谱柱为:x2(4.6×250mm,10μm,14011407y)。进一步地,步骤(2)所述正向制备色谱柱中流动相流速为1ml/min,柱温为30℃,波长为277nm。进一步地,步骤(2)所述分段收集是从45min开始,每1min收集1次。本发明的替西罗莫司分离纯化方法,在替西罗莫司合成过程中,保证高转化率的条件下,对关键中间体——酯化物2,2,5-三甲基[1,3-二氧六烷]-5-羧酸-42-酯-雷帕霉素进行硅胶纯化,降低了后续替西罗莫司的纯化难度;采用正向制备色谱柱分离纯化更易于浓缩干燥,能有效的降低替西罗莫司产品中异构体的含量;以正庚烷/乙醇进行梯度洗脱可使样品中的大极性及低极性的杂质充分分离,提高替西罗莫司产品的纯度。本发明的替西罗莫司分离纯化方法生成的替西罗莫司产品纯度高达99%,收率高于80%,异构体含量低于0.2%。通过本发明的分离纯化方法得到的替西罗莫司各项指标均优于现有技术中得到的产品,且本发明方法操作简便,适合工业化生产。显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。附图说明图1纯化时hplc谱图图2产品纯度检测图谱及数据(合并纯度≥98%洗脱液)具体实施方式下面以实施例对本发明做进一步的阐述和说明。实施例1本发明替西罗莫司的分离纯化a.将2,2,5-三甲基-5-羧酸-1,3-二氧六烷溶解于正己烷中,于25±5℃下加入二氯亚砜,混合后质量体积比m2,2,5-三甲基-5-羧酸-1,3-二氧六烷:v正己烷:m二氯亚砜=1g:14ml:1.37g,搅拌15h。减压浓缩至干,得无色油状液体,即酰氯。b.将雷帕霉素、n,n-二异丙基乙胺(dipea)与二氯甲烷(dcm)混合,其质量体积比m雷帕霉素:vdipea:vdcm=1g:0.72ml:2.83ml,0-5℃下将上述a中制备的酰氯滴入,使酰氯浓度为1g/ml,25±5℃下搅拌反应22h后取样,hplc检测雷帕霉素含量小于0.5%后,浓缩蒸干,所得粗品以石油醚:乙酸乙酯1:1为洗脱剂,通过200-300目层析柱硅胶纯化,等度洗脱得酯化物2,2,5-三甲基[1,3-二氧六烷]-5-羧酸-42-酯-雷帕霉素。c.将2,2,5-三甲基[1,3-二氧六烷]-5-羧酸-42-酯-雷帕霉素溶解于四氢呋喃(thf)中,25±5℃下滴加2mol/l硫酸水溶液,使体系中各成分的质量体积比为m2,2,5-三甲基[1,3-二氧六烷]-5-羧酸-42-酯-雷帕霉素:vthf:v硫酸水=1g:7.95ml:1.88ml,反应14h。饱和碳酸钠水溶液调节ph至7-8,乙酸乙酯(ea)萃取,无水硫酸钠干燥,减压浓缩得到替西罗莫司粗品。(2)用乙醇-正庚烷(体积比1:1)溶解替西罗莫司粗品,得粗品浓度0.2g/ml,采用正向制备色谱柱x2(4.6×250mm,10μm,14011407y),流速为1ml/min,柱温为30℃,波长为277nm。用乙醇-正庚烷洗脱(0-25min内正庚烷/乙醇体积比由1:0升至1:1梯度洗脱,25-70min为正庚烷/乙醇体积比为1:1等度洗脱,70-90min内正庚烷/乙醇体积比由1:1升至1:4梯度洗脱),分段收集洗脱液(见图1,表1),经hplc检测后,合并替西罗莫司纯度≥98%的洗脱液(见图2,表2),减压干燥得到替西罗莫司纯品。前述hplc检测方法为:以辛烷基硅烷键合硅胶为填充柱(agilentzorbaxsb-c8,4.6mm×150mm,3.5μm),流动相a为水,流动相b为甲醇,流速为0.8ml/min,柱温35℃,检测波长为277nm;梯度洗脱程序如下:表1洗脱液收集馏分时间馏分时间馏分时间f145.7~46.7f953.7~54.7f1761.7~62.7f246.7~47.7f1054.7~55.7f1862.7~63.7f347.7~48.7f1155.7~56.7f1963.7~64.7f448.7~49.7f1256.7~57.7f2064.7~65.7f549.7~50.7f1357.7~58.7f2165.7~66.7f650.7~51.7f1458.7~59.7f2266.7~67.7f751.7~52.7f1559.7~60.7f2367.7~68.7f852.7~53.7f1660.7~61.7表2洗脱液纯度数据通过本发明分离纯化方法得到的替西罗莫司纯品收率80.1%,经前述hplc法检测所得替西罗莫司纯品单杂小于0.5%,主体与异构体比为99.4:0.2。本发明方法具有高转化率,降低了成本,还降低了替西罗莫司的纯化难度,得到的替西罗莫司产品中异构体含量低,大极性及低极性杂质少,产品纯度高,提高了质量,具有广阔的市场应用前景。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1