巯基改性高分子化合物及其制备方法和用途与流程
1.本发明属于高分子材料领域,具体涉及一种巯基改性高分子化合物及其制备方法和用途。
背景技术:
2.生物相容性高分子具有很多重要的生理功能,常见的生物相容性高分子包括:多糖、蛋白质以及合成型高分子等。其中典型的天然生物相容性高分子为透明质酸,1934年,karl mayer教授首次从牛眼玻璃体中提取获得天然透明质酸,天然透明质酸是由d-葡糖醛酸和n-乙酰基-d-葡糖胺的交替单元组成的天然杂多糖。其后经过数十年的研究,人们发现其在人和其他脊椎动物的结缔组织中广泛存在,例如细胞间隙、运动关节组织、脐带、皮肤、软骨、血管壁、滑液以及鸡冠等组织和器官中都含有透明质酸。透明质酸属于线性高分子多糖,结构中含有二糖重复单元,重复单元中的d-葡萄糖醛酸通过β-1,3糖苷键与n-乙酰基-d-葡糖胺连接,成千上万的二糖重复单元通过β-1,4糖苷键相连,形成整个高分子直链、线性结构。在人体生理状态下,透明质酸通常以钠盐的形式存在。其中,透明质酸钠及其凝胶在骨科、妇科及整形科等领域应用广泛,还可用于作为眼用制剂载体或直接作为眼用制剂应用于眼科手术,即透明质酸钠类产品在眼科手术中也有重要应用。透明质酸钠还是关节滑液及软骨的重要组成成分,通过提高关节内透明质酸钠的含量,可以增加关节滑液的粘稠性和润滑功能,发挥保护软骨、促进关节愈合和再生、缓解疼痛、增加关节活动度等作用。且有文献报道:大量的动物实验和临床应用表明,在预防和降低妇产科手术造成的粘连方面,透明质酸及其钠盐是一种安全、有效的理想物质;其中,透明质酸钠的水溶液,是一种非牛顿型流体,有着良好的粘弹性和流变性,而且一般而言,低浓度的透明质酸溶液主要表现出粘性,高浓度的透明质酸溶液主要表现出弹性,因此可以根据实际使用的需求调整其浓度。
3.天然的透明质酸或其钠盐虽然应用领域广泛且具有多种明确的应用优势,但天然的透明质酸或其钠盐也有明确的缺点。首先,天然的透明质酸或其钠盐在体内的半衰期短,在生物体内降解时间一般不多于7天,造成半衰期短的主要原因为天然的透明质酸或其钠盐的平均分子量较小,且具有较好的流动性,容易分散于组织中并被吸收和代谢,其直接表现为在溶液态是低粘度的。其次,天然的透明质酸或其钠盐具有稳定性较差,易发生降解的缺点。第三,天然的透明质酸或其钠盐具有亲水性过强的缺点。为了克服天然的透明质酸或其钠盐的上述缺点,现有技术中采用的常见手段为化学改性和分子间交联,化学改性和分子间交联也已成为研究的热点方向之一。
4.生物相容性高分子的改性研究是近年来对多糖、蛋白质及合成型高分子等生物相容性高分子研究的重点方向之一。其中,生物相容性高分子的化学改性即为该类化合物的结构修饰与改造,主要包括以下几种类型:1、疏水性改性,代表例为烷基化改性;2、羧基化改性,代表例为羧甲基化改性;3、巯基化改性;4、接枝化改性,以透明质酸的接枝反应为例,其是将小分子物质或聚合物接枝到透明质酸的主链上,代表例为透明质酸与高密度聚乙烯
接枝共聚。其中,巯基化改性的生物相容性高分子因其具有易交联形成水凝胶、抗氧化等特点,十分适合抗氧化保健品、生物医药、医疗美容整形以及化妆品等领域的应用。生物相容性高分子的巯基化改性过程,一般是指引入自由巯基的化学改性过程,通常多糖、蛋白质和合成型高分子等的功能性基团,如:羧基、氨基、羟基等,可以通过适当的化学反应引入自由巯基。
5.现有的巯基改性高分子化合物的制备,尤其是巯基改性生物相容性高分子化合物的制备,以巯基化改性透明质酸为例,主要有以下方法:
6.prestwich与舒晓正等人首次报道了二硫代二酰肼法合成的巯基透明质酸衍生物,以下简称ha-sh,结构见图1(文献1:biomacromolecules 2002,3,1304和公开号为cn101511875a的专利文献)。该方法需要通过多步反应预先合成含有二硫键的二酰肼化合物,并且反应过程中使用的试剂毒性比较高,例如水合肼就属于高毒性化合物,其大鼠经口口服的ld
50
为129mg/kg。随后通过与edci,即碳化二亚胺脱水剂,缩合反应使透明质酸与二硫代二酰肼化合物交联形成凝胶,该步骤由于反应体系整体凝胶化,无法持续搅拌以维持ph4.75的最佳反应条件,因此不易得到均一化的巯基衍生物。所得凝胶经过二硫苏糖醇还原反应得到巯基修饰的透明质酸衍生物。
7.prestwich等人于2008年报道了一种硫代环乙烷修饰合成ha-sh的方法,具体见图2(文献2:biomaterials 2008,29,1388)。该方法不需要预先合成修饰试剂,但是受限于透明质酸伯醇的低反应活性,其修饰比例比较低。
8.2007年,shimobouji等人报道了一种涉及二酰肼和traut’s试剂的两步修饰法合成ha-sh,具体见图3(文献3:j biomed mater res a.2007,80,916.)。该方法第一步使用过量的二酰肼化合物来修饰透明质酸,中间产品经过透析冻干等纯化步骤得到酰肼修饰的透明质酸衍生物。第二步使用traut’s试剂来修饰酰肼透明质酸衍生物,经过一系列纯化操作得到巯基修饰的透明质酸衍生物。该反应第二步所用traut’s试剂价格昂贵,参考价为:374€/g,sigma公司,纯度98%。这极大地阻碍了该方法的大规模工业化生产。
9.2007年,tae gwan park等人报道了一种使用光胺修饰的方法合成ha-sh,具体见图4(文献4:journal of controlled release 2007,119,245)。该方法与二硫代二酰肼法类似,使用含有二硫键的光胺作为修饰原料,通过edci脱水缩合,二硫苏糖醇还原二硫键,最终得到ha-sh衍生物。由于氨基反应活性低于酰肼反应活性,因此该方法取代度低于二硫代二酰肼法合成的ha-sh。
10.2012年,c.yan等人报道了使用β-巯基乙胺来修饰透明质酸合成ha-sh的方法,具体见图5(文献5:acta biomaterialia 2012,8,3643)。该方法直接使用含有硫醇的小分子化合物来修饰透明质酸的羧基,反应过程对硫醇基团不采取保护措施。由于硫醇同样可以和羧基发生edci催化偶联反应,因此该方法所得产品实际为巯基和胺基修饰透明质酸的混合物,并且硫醇取代度远远低于酰肼法合成的ha-sh。
11.2015年,andreas bernkop-schunrch等人报道了一种使用半胱氨酸乙酯的方法合成ha-sh,具体见图6(文献6:international journal of pharmaceutics 2015,478,383)。该方法与β-巯基乙胺法类似,在反应过程中的硫醇也会参与羧基偶联反应,取代效率无法有效提高。
12.公开号为cn101200504a的专利文献中披露了一种使用改性二硫代二酰肼化合物
修饰透明质酸的ha-sh合成方法,具体见图7。该方法所使用改性二硫代二酰肼化合物拥有更长的骨架结构,因此所得ha-sh中巯基的反应活性比普通二硫代二酰肼法得到的硫醇活性高。但是该改性二硫代二酰肼需要多步合成法来制备,工业化成本比较高。
13.公开号为cn103613686a的专利文献中披露了一系列ha-sh的合成方法,其中包含了普通二硫代二酰肼法(即文献1和专利cn101511875a)中的ha-sh结构,具体见图8。
14.上述方法制备得到的巯基改性高分子化合物,可以归纳为以下三种类型:第一种,对高分子化合物的侧链羧基进行酰胺化修饰加成含有巯基的小分子片段;第二种,对高分子化合物的侧链羧基进行酰胺化或酰肼化修饰后进行二次功能化通过还原反应或开环反应得到含有巯基的侧链结构;第三种,对高分子化合物的侧链羟基进行高碱性条件下的环硫乙烷开环反应得到含有巯基的侧链结构。这些方法存在以下缺陷:1)反应试剂较为昂贵,如edci、traut’s试剂等;2)反应过程涉及非商业化试剂的使用,如二硫代二酰肼化合物需要额外两步化学合成来制备,大大增加了工业化成本;3)针对羧基改性的方法需要使用edci,其最佳反应ph为4.75,会引起某些高分子化合物(如透明质酸)的不可逆降解;4)针对羟基的环硫乙烷改性方法涉及到ph=10的苛刻反应条件,同样会引起某些高分子化合物的不可逆降解;5)针对氨基修饰的方法受限于反应活性较低,巯基修饰程度普遍不高。
15.如上所述,为了克服天然生物相容性高分子的缺点,现有技术中还可以采用分子间交联的手段,具体而言,生物相容性高分子化合物以分子间交联的方式可以改变其理化性质,例如将透明质酸钠通过交联剂作用制得交联透明质酸钠凝胶,该凝胶作为填充物注入体内具有填充效果好且生物相容性高等优点,已经被广泛用于医疗和美容领域。分子间交联中采用的交联剂一般为环氧化物交联剂,包括但不限于以下范围:1,4-丁二醇二缩水甘油醚(bdde)、乙二醇二缩水甘油醚(egdge)、1,6-己二醇二缩水甘油醚、丙二醇二缩水甘油醚、聚(丙二醇)二缩水甘油醚、聚(四亚甲撑二醇)二缩水甘油醚、新戊二醇二缩水甘油醚、聚甘油聚缩水甘油醚、二甘油聚缩水甘油醚、甘油聚缩水甘油醚、三羟甲基丙烷聚缩水甘油醚、1,2-(双(2,3-环氧丙氧基)乙烯、季戊四醇聚缩水甘油醚或山梨糖醇聚缩水甘油醚等,其中应用最广的是1,4-丁二醇二缩水甘油醚(bdde)。
16.现有技术中,生物相容性高分子(如透明质酸或其钠盐)的交联化处理的改造方式的最主要缺点是:一方面是交联剂的加入带来了不可避免的交联剂残留,而残留的交联剂往往具有毒性,进而影响到交联化生物相容性高分子(如交联透明质酸或其钠盐)在应用、尤其是在人体的应用中带来的毒性反应或不良反应;另一方面是为了纯化小分子交联剂而带来的复杂工艺过程,使交联化生物相容性高分子(如交联透明质酸或其钠盐)的生产成本居高不下。
技术实现要素:
17.为解决上述问题,本发明的目的在于提供一种新结构的巯基改性的生物相容性高分子系列化合物,所述巯基改性的生物相容性高分子系列化合物具有诸多有益特点,使得其更加利于用于抗氧化保健品、生物医药、医疗美容整形以及化妆品等领域。
18.本发明的第二个目的在于提供一种制备上述巯基改性的生物相容性高分子系列化合物的方法,该方法克服了现有技术中存在的诸多缺点和不足,极具应用前景。具体的,所述方法克服了现有技术中存在的以下缺点和不足:1)修饰试剂需要经过多步反应合成,
如:二硫代二酰肼法和改性二硫代二酰肼法;2)修饰试剂合成过程中需要应用水合肼等高毒性有机物,不符合绿色化学或环境友好型社会的发展需求;3)涉及到edci偶联剂的巯基修饰方法在活化阶段需要维持反应体系ph=4.75,进入凝胶阶段后无法通过搅拌等均质化作用实现反应体系稳定均一,工业化操作难度大;4)ethylene sulfide法合成ha-sh需要在ph=10条件下进行,无法避免透明质酸在合成过程中发生的降解反应;另外,硫代环乙烷为易燃有毒危险品,在水中溶解度较低,限制了该方法获得较高取代度的可能;5)traut’s试剂酰肼透明质酸衍生物修饰法受限于高昂的原料成本,无法大规模工业化生产;6)由于硫醇与氨基都属于活性较高亲核试剂,因此β-巯基乙胺与半胱氨酸乙酯等硫醇直接修饰过程存在氨基与羧基的缩合主反应以及硫醇与羧基缩合的副反应,且该副反应难以通过改变反应条件来完全避免。
19.本发明第一方面是提供一种巯基改性高分子化合物,被改性的高分子化合物的结构上含有-cooh、-nh2、-oh、式a所示丙烯酸酯类基团、式b所示丙烯酰胺类基团、式c所示丙烯酰类基团中的至少一种,
[0020][0021]
其中,所述-cooh和/或-nh2和/或-oh和/或丙烯酸酯类基团和/或丙烯酰胺类基团和/或丙烯酰类基团的部分或全部被修饰形成端基为下述基团的侧链:
[0022][0023]
上述基团中,*表示连接点;
[0024]
r1选自氢,卤素,脂肪基团,芳香基团等;
[0025]
r2和r3相同或不同,彼此独立地选自氢,卤素,脂肪基团,芳香基团等;
[0026]
r4为多巯基化合物片段。
[0027]
本发明第二方面是提供一种上述巯基改性高分子化合物的制备方法,其包括以下步骤:
[0028]
1)结构上含有-cooh、-nh2、-oh中的至少一种的高分子化合物的丙烯酰化步骤,即将高分子化合物的结构上含有的-cooh、-nh2、-oh的至少一种直接或间接与如下基团连接:
[0029][0030]
r1、r2和r3的定义同前,*表示连接点;
[0031]
或者,结构上含有上述式a所示丙烯酸酯类基团、上述式b所示丙烯酰胺类基团、上述式c所示丙烯酰类基团中至少一种的高分子化合物直接作为反应原料;
[0032]
2)将步骤1)得到的高分子化合物的至少一种与多巯基化合物hs-r
4-sh反应,r4的定义同前,制备得到所述的巯基改性高分子化合物。
[0033]
本发明的第三方面是提供一种上述巯基改性高分子化合物的用途,其用于抗氧化
保健品,生物医药,医疗美容整形,以及化妆品(如抗氧化类化妆品、保水补湿类化妆品中的至少一种)等领域。
[0034]
本发明的有益效果
[0035]
本发明制备的经过硫醇化改造的多糖、蛋白质、合成型高分子等生物相容性高分子化合物,除了可预料或可推测的硫醇含量提高、主链分子的分子量变化很小之外,令人意想不到的是硫醇化修饰过的各生物相容性高分子化合物的产物,在粘度、保水性、抗氧化性能等方面对比修饰前的分子具有显著差别和明显提高。而粘度、保水性、抗氧化性能是多糖、蛋白质、合成型高分子等生物相容性高分子化合物的主要理化性质,而这几项理化性质与上述生物相容性高分子化合物的应用范围密切相关。具体的,在表征本发明所述的系列生物相容性高分子化合物的物理化学特征时,粘度是最主要的性能指标,除了粘度本身是反应分子量大小的外在指标之外,也是影响各生物相容性高分子化合物在人体内治疗或整形等效果的关键指标,粘度大则更难以在组织中分散,由此减少组织对此化合物的吸收速度,进而使生物相容性高分子化合物在人体内保持更长久的作用时间,即提高其在体内的代谢半衰期。而以透明质酸为代表的各类生物相容性高分子化合物,保水补湿功能是其应用于临床、化妆品等的重要性功能指标,保水率是评价生物相容性高分子化合物的重要指标和优效对比指标。抗氧化性能是本发明所涉及的生物相容性高分子化合物中的多种代表性化合物的主要功能指标,也是这些生物相容性高分子化合物应用于抗氧化类化妆品、抗氧化保健品、抗氧化医疗用品的主要功能指标,而抗氧化性能测定已具备完善的检测方法和评价体系。
[0036]
具体而言,本发明提供了一种新结构的巯基改性的生物相容性高分子系列化合物,该系列新化合物是由本发明中所提及的各种带有硫醇基团的侧链经化学合成反应修饰的各种生物相容性高分子化合物,该系列新化合物在粘度、保水性、抗氧化性能等诸多的材料性能方面具有明显优势,具体的具有以下特点:
[0037]
1.由生物相容性高分子通过合成反应在活性位点键合上新的侧链而产生的新化合物结构。
[0038]
2.合成过程中,对比被修饰前的化合物合成过程,对比现有技术中的最接近化合物的合成过程,具有的优势包括:1)该反应温和可控,在中性条件下反应避免了高分子化合物主链在苛刻酸碱度环境中的降解;2)硫醇与共轭双键的迈克尔加成反应效率极高,且没有任何副产物生成,符合原子经济学原理和绿色化学发展条件;3)多巯基化合物(如:二巯基,三巯基,四巯基甚至巯基数量更多的多巯基化合物)选自商业化商品,选择广泛,无需经过多步合成反应预先制备巯基修饰试剂,工业化成本低;4)所述多巯基化合物的结构灵活可调,侧链片段长短可以根据实际需求订制化改性,从而实现对巯基侧链的活性控制以及微观交联三维结构孔道大小以及孔隙率的自由调节;5)现有技术中的巯基修饰方法的理论巯基取代度最高仅为100%(针对有重复单元的高分子化合物),本发明通过三巯基以及四巯基化合物的使用,可以使理论巯基取代度分别达到200%以及300%,该效果是其他现有技术的方法所不具备的独有优势。
[0039]
3.相对于结构改造前、或相对于现有技术中最接近的改性后的生物相容性高分子化合物,本发明的新结构的系列化合物具备意想不到的有益技术效果并明显区别于现有技术,至少具有不同的且意料不到的物理化学性质。
[0040]
4.由所述新的材料性质而带来的在潜在应用领域的优势,或具有潜在的新用途。
[0041]
基于此,所述巯基改性的高分子系列化合物更加利于用于抗氧化保健品、生物医药、医疗美容整形以及化妆品等领域。
[0042]
进一步的,本发明还提供一种制备上述巯基改性高分子化合物的方法,具体的,本发明使用(甲基)丙烯酸酯类化合物修饰的高分子化合物与多巯基化合物通过硫醇与共轭双键的迈克尔加成反应来制备巯基改性高分子化合物,该制备方法除了能实现本发明中所述的新化合物结构目标之外,还具以下优势:能够灵活有效地控制合成产物的结构、组成、大量化合物分子末端功能基团的种类与含量等;采用生物相容性高的试剂,有效控制生产成本并降低合成工艺过程中的毒性;能够保证在采用安全性试剂和简单反应步骤的条件下,获得原材料结构和生物活性保持良好、功能基团种类和含量可按需调控的可作为细胞外基质材料巯基改性的生物相容性高分子系列化合物,满足多种临床应用要求。具体而言,所述方法具备以下优势:
[0043]
1)该反应温和可控,在中性条件下即可实现修饰反应,避免了高分子化合物主链在苛刻酸碱度环境中的降解;
[0044]
2)该硫醇与共轭双键的迈克尔加成反应效率极高,且没有任何副产物生成,符合原子经济学原理和绿色化学发展条件;
[0045]
3)多巯基化合物(如,二巯基,三巯基,四巯基甚至巯基数量更多的多巯基化合物)选自商业化商品,选择广泛,无需经过多步合成反应预先制备巯基修饰试剂,工业化成本低;
[0046]
4)所述多巯基化合物的结构灵活可调,侧链片段长短可以根据实际需求订制化改性,从而实现对巯基侧链的活性控制以及微观交联三维结构孔道大小以及孔隙率的自由调节;
[0047]
5)现有技术中的巯基修饰方法的理论巯基取代度最高仅为100%(针对有重复单元的高分子化合物),本发明通过三巯基以及四巯基化合物的使用,可以使理论巯基取代度分别达到200%以及300%,该效果是其他现有技术的方法所不具备的独有优势。
附图说明
[0048]
图1文献1中报道的主要反应过程(二酰肼法合成ha-sh);
[0049]
图2文献2中报道的主要反应过程(ethylene sulfide法合成ha-sh);
[0050]
图3文献3中报道的主要反应过程(酰肼后修饰法合成ha-sh);
[0051]
图4文献4中报道的主要反应过程(光胺修饰法合成ha-sh);
[0052]
图5文献5中报道的主要反应过程(β-巯基乙胺法合成ha-sh);
[0053]
图6文献6中报道的主要反应过程(半胱氨酸乙酯修饰合成ha-sh);
[0054]
图7文献cn101200504a中报道的主要反应过程(二酰肼法合成ha-sh);其中,r1和r2是亚烷基、取代亚烷基、芳香基、聚醚等(注:此处的r1和r2的定义仅限于图7和文献cn101200504a中);p是指侧链含有羧基的高分子化合物残基;
[0055]
图8文献cn103613686a中报道的主要反应过程(与文献1类似,二酰肼法合成ha-sh);
[0056]
图9实施例1的反应方程式;
[0057]
图10实施例2的反应方程式;
[0058]
图11实施例3的反应方程式;
[0059]
图12实施例4的反应方程式;
[0060]
图13实施例5的反应方程式;
[0061]
图14实施例6的反应方程式;
[0062]
图15实施例7的反应方程式;
[0063]
图16实施例8的反应方程式;
[0064]
图17实施例9的反应方程式;
[0065]
图18实施例10的反应方程式;
[0066]
图19实施例11的反应方程式;
[0067]
图20实施例12的反应方程式;
[0068]
图21实施例13的反应方程式(其中的i=10-90%,j=10-90%,i2+i3=i,j2+j3=j,h=j,i+j=100%,k1=1-1000);
[0069]
图22实施例14的反应方程式;
[0070]
图23实施例15的反应方程式;
[0071]
图24实施例16的反应方程式;
[0072]
图25实施例17的反应方程式;
[0073]
图26实施例18的反应方程式;
[0074]
图27实施例19的反应方程式;
[0075]
图28实施例20的反应方程式;
[0076]
图29实施例21的反应方程式;
[0077]
图30实施例22的反应方程式;
[0078]
图31实施例23的反应方程式;
[0079]
图32实施例24的反应方程式;
[0080]
图33实施例25的反应方程式;
[0081]
图34实施例26的反应方程式;
[0082]
图35实施例27的反应方程式;
[0083]
图36实施例28的反应方程式;
[0084]
图37ha-a1的结构式和其1h-nmr谱图;
[0085]
图38ha-a2的结构式和其1h-nmr谱图;
[0086]
图39ha-ma1的结构式和其1h-nmr谱图;
[0087]
图40ha-ma2的结构式和其1h-nmr谱图;
[0088]
图41chs-a的结构式和其1h-nmr谱图;
[0089]
图42chs-ma的结构式和其1h-nmr谱图;
[0090]
图43gelatin-a的结构式和其1h-nmr谱图;
[0091]
图44gelatin-ma的结构式和其1h-nmr谱图;
[0092]
图45cts-a的结构式和其1h-nmr谱图;
[0093]
图46cts-ma的结构式和其1h-nmr谱图;
[0094]
图47phema-a的结构式和其1h-nmr谱图;
[0095]
图48phema-ma的结构式和其1h-nmr谱图;
[0096]
图49pva-a的结构式和其1h-nmr谱图;
[0097]
图50pva-ma的结构式和其1h-nmr谱图;
[0098]
图51ha-a1-sh1的结构式和其1h-nmr谱图;
[0099]
图52ha-a2-sh1的结构式和其1h-nmr谱图;
[0100]
图53ha-ma1-sh1的结构式和其1h-nmr谱图;
[0101]
图54ha-ma2-sh1的结构式和其1h-nmr谱图;
[0102]
图55chs-a-sh1的结构式和其1h-nmr谱图;
[0103]
图56chs-ma-sh1的结构式和其1h-nmr谱图;
[0104]
图57gelatin-a-sh1的结构式和其1h-nmr谱图;
[0105]
图58gelatin-ma-sh1的结构式和其1h-nmr谱图;
[0106]
图59cts-a-sh1的结构式和其1h-nmr谱图;
[0107]
图60cts-ma-sh1的结构式和其1h-nmr谱图;
[0108]
图61phema-a-sh1的结构式和其1h-nmr谱图;
[0109]
图62phema-ma-sh1的结构式和其1h-nmr谱图;
[0110]
图63hb-peg-sh1的结构式和其1h-nmr谱图;
[0111]
图64pva-a-sh1的结构式和其1h-nmr谱图;
[0112]
图65pva-ma-sh1的结构式和其1h-nmr谱图;
[0113]
图66ha-a1-sh2的结构式和其1h-nmr谱图;
[0114]
图67ha-a1-sh3的结构式和其1h-nmr谱图;
[0115]
图68ha-a2-sh2的结构式和其1h-nmr谱图;
[0116]
图69ha-a2-sh3的结构式和其1h-nmr谱图;
[0117]
图70ha-a2-sh4的结构式和其1h-nmr谱图;
[0118]
图71ha-a2-sh5的结构式和其1h-nmr谱图;
[0119]
图72ha-a2-sh6的结构式和其1h-nmr谱图;
[0120]
图73ha-a2-sh7的结构式和其1h-nmr谱图;
[0121]
图74ha-a2-sh8的结构式和其1h-nmr谱图;
[0122]
图75ha-ma1-sh5的结构式和其1h-nmr谱图;
[0123]
图76ha-ma1-sh6的结构式和其1h-nmr谱图;
[0124]
图77ha-ma2-sh7的结构式和其1h-nmr谱图;
[0125]
图78ha-ma2-sh8的结构式和其1h-nmr谱图;
[0126]
图79ellman检测标准曲线;
[0127]
图80dpph自由基含量检测标准曲线;
[0128]
图81dpph自由基捕获百分比。
具体实施方式
[0129]
[巯基改性高分子化合物]
[0130]
如前所述,本发明提供了一种巯基改性高分子化合物,被改性的高分子化合物的结构上含有-cooh、-nh2、-oh、式a所示丙烯酸酯类基团、式b所示丙烯酰胺类基团、式c所示
丙烯酰类基团中的至少一种,
[0131][0132]
其中,所述-cooh和/或-nh2和/或-oh和/或丙烯酸酯类基团和/或丙烯酰胺类基团和/或丙烯酰类基团的部分或全部被修饰形成端基为下述基团的侧链:
[0133][0134]
上述基团中,*表示连接点;
[0135]
r1选自氢,卤素,脂肪基团,芳香基团等;具体的,所述卤素、脂肪基团、芳香基团满足下文中的进一步的定义;优选地,r1选自氢,卤素,脂肪基团;还优选地,r1选自氢,卤素,c1-6烷基(例如甲基、乙基等);
[0136]
r2和r3相同或不同,彼此独立地选自氢,卤素,脂肪基团,芳香基团等;具体的,所述卤素、脂肪基团、芳香基团满足下文中的进一步的定义;
[0137]
r4为多巯基化合物片段。
[0138]
在一个具体的实施方式中,所述-cooh和/或-nh2和/或-oh和/或丙烯酸酯类基团和/或丙烯酰胺类基团和/或丙烯酰类基团的部分或全部被修饰形成以下结构的至少一种:
[0139][0140][0141]
上述结构中,r选自亚烃基、亚芳基、酰胺残基、酰肼残基等;*表示连接点;1*表示与r的左侧基团的连接点;2*表示与r的右侧基团的连接点;r1、r2、r3和r4的定义同前。
[0142]
其中,所述-cooh、-nh2、-oh、式a所示丙烯酸酯类基团、式b所示丙烯酰胺类基团、式c所示丙烯酰类基团中的至少一种可以是直接连接在高分子化合物的主链上,也可以是通过r’基团连接在高分子化合物的主链上,所述r’可以是含有杂原子的基团、亚烃基、亚芳基或下述连接基团:
[0143][0144]
上式中,r”是亚烃基或亚芳基,n’为1-1000的整数,*表示连接点。
[0145]
其中,所述含有杂原子的基团包括但不限于:酯基、酰胺残基或酰肼残基。具体的,所述酯基、酰胺残基或酰肼残基满足下文中的进一步的定义。
[0146]
其中,所述被改性的高分子化合物包含天然粘多糖聚合物,如壳聚糖类(具体可以是壳聚糖、乙二醇壳聚糖、羧甲基壳聚糖等),硫酸软骨素,透明质酸,海藻酸盐等中的至少一种;蛋白质,如明胶、纤维蛋白、血清蛋白等;和/或,合成聚合物,如聚乙烯醇,聚(甲基)丙烯酸,聚(甲基)丙烯酸羟烷基酯(例如聚(甲基)丙烯酸羟乙酯等),超支化聚乙二醇等中的至少一种。
[0147]
其中,用ellman法检测的所述巯基改性高分子化合物的巯基含量为0.01-30mmol/g,例如为0.1-10.0mmol/g,还例如为0.3-5.0mmol/g,再例如为0.5-3.0mmol/g。
[0148]
其中,所述巯基改性高分子化合物的分子量与改性前高分子化合物的分子量基本不变。
[0149]
例如,本发明的巯基改性高分子化合物包括如下结构的至少一种:
[0150]
[0151]
[0152][0153]
上述结构中,a为所述结构上含有至少一个-cooh、-nh2、-oh、式a所示丙烯酸酯基团、式b所示丙烯酰胺基团、式c所示丙烯酰类基团的被改性高分子化合物的片段;r、r’、r1、
r2、r3和r4的定义同前;(n2+n3)/(n1+n2+n3)表示丙烯酰化度;n3/(n1+n2+n3)表示巯基化程度,与上述的用ellman法检测的所述巯基改性高分子化合物的巯基含量是对应的;所述n1可以为0,若为0,则不用限定丙烯酰化度,仅是n3/(n2+n3)表示巯基化程度,与上述的用ellman法检测的所述巯基改性高分子化合物的巯基含量是对应的;所述n2可以为0,若为0,则n3/(n1+n3)既表示丙烯酰化度,又表示巯基化程度,与上述的用ellman发检测的所述巯基改性高分子化合物的巯基含量是对应的。
[0154]
具体的,所述a可以是如下所示结构:
[0155][0156]
上述各结构中,*表示主链重复单元之间的连接点;**表示-cooh、-nh2、-oh、式a所示丙烯酸酯类基团、式b所示丙烯酰胺类基团、式c所示丙烯酰类基团与上述片段之间的连接点、或者通过r’基团与上述片段之间的连接点。
[0157]
所述a还可以是下述聚合物gelatin-a、gelatin-ma、cts-a、cts-ma、phema-a、phema-ma、hb-peg、pva-a、pva-ma、chs-a或chs-ma中的脱除含丙烯酰侧链后的剩余的片段或重复单元:
[0158][0159][0160]
需要说明的是,gelatin-a、gelatin-ma、cts-a、cts-ma、phema-a、phema-ma、hb-peg、pva-a、pva-ma、chs-a或chs-ma分别是具有上述结构的聚合物名称的简写,其中的字母分开后不与本发明中其他部分出现的字母含义相关。
[0161]
如前所述,r4为多巯基化合物片段,例如,所述-s-r
4-sh片段可以由下述但不仅限于下述多巯基化合物引入:
[0162][0163]
其中,n4=2-30的整数,例如n=2、3、4、5或6等;n5=1-30的整数,例如为1、2、3、4、5等;n6=1-30的整数,例如为1、2、3、4、5等;
[0164]
4-arm-peg-sh表示含有四个巯基团的peg聚合物;6-arm-peg-sh表示含有六个巯基基团的peg聚合物;8-arm-peg-sh表示含有八个巯基的peg聚合物;所述peg是聚乙二醇的缩写。
[0165]
本发明中若没有特殊说明,其中出现的n、n’、n1、n2、n3、n4、n5、n6、m1、m2、i、j、k1、h均是指结构式中出现的重复单元的个数。其取值范围属于本领域已知的常规范围。
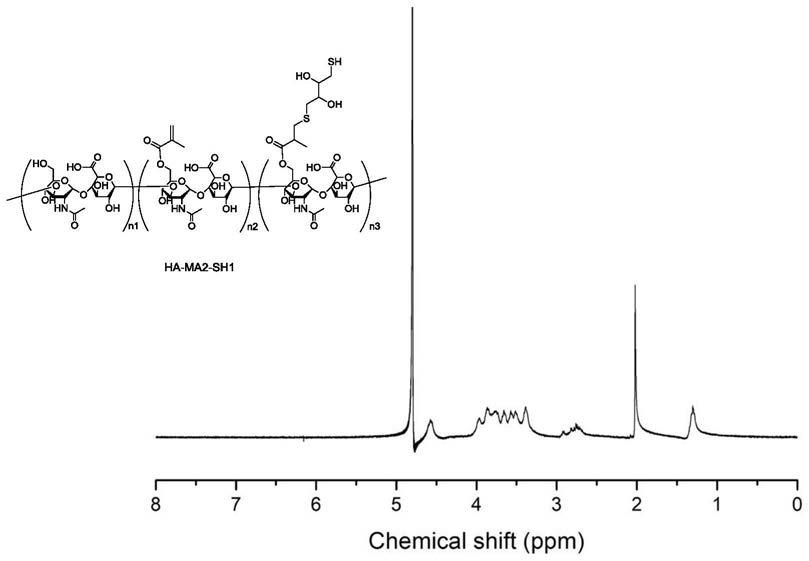
[0166]
如前所述,r1选自氢,卤素,脂肪基团,芳香基团等;r2和r3相同或不同,彼此独立地选自氢,卤素,脂肪基团,芳香基团等。
[0167]
如前所述,所述r可选自亚烃基、亚芳基、酰胺残基、酰肼残基等。
[0168]
如前所述,所述r’可选自含有杂原子的基团、亚烃基、亚芳基等。
[0169]
如前所述,所述r”可选自亚烃基、亚芳基等。
[0170]
所述卤素是指氟、氯、溴或碘。
[0171]
所述脂肪基团例如为直链或支链饱和/不饱和脂肪基团,具体的可以是烷基、烯基或炔基。
[0172]
本发明单独使用或用作后缀或前缀的“烃基”例如为直链或支链饱和/不饱和脂肪基团,具体的可以是烷基、烯基或炔基。
[0173]
本发明单独使用或用作后缀或前缀的“烷基”意在包括具有1至20个,优选1-6个碳原子的支链和直链饱和脂族烃基。例如,“c
1-6
烷基”表示具有1、2、3、4、5或6个碳原子的直链和支链烷基。烷基的实例包括但不限于甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、戊基和己基。
[0174]
本发明单独使用或用作后缀或前缀的“烯基”意在包括具有2至20个,优选2-6个碳原子(或若提供了碳原子的具体数目,则指该具体数目)的包含烯基或烯烃的支链和直链脂族烃基。例如,“c
2-6
烯基”表示具有2、3、4、5或6个碳原子的烯基。烯基的实例包括但不限于
乙烯基、烯丙基、1-丙烯基、1-丁烯基、2-丁烯基、3-丁烯基、2-甲基丁-2-烯基、3-甲基丁-1-烯基、1-戊烯基、3-戊烯基和4-己烯基。
[0175]
本发明单独使用或用作后缀或前缀的“炔基”意在包括具有2至20个,优选2-6个碳原子(或若提供了碳原子的具体数目,则指该具体数目)的包含炔基或炔烃的支链和直链脂族烃基。例如乙炔基、丙炔基(例如l-丙炔基、2-丙炔基)、3-丁炔基、戊炔基、己炔基和1-甲基戊-2-炔基。
[0176]
所述芳香基团指由5至20个碳原子构成的芳族环结构。例如:包含5、6、7和8个碳原子的芳族环结构可以是单环芳族基团例如苯基;包含8、9、10、11、12、13或14个碳原子的环结构可以是多环的例如萘基。芳环可在一个或多个环位置取代有取代基,所述取代基为烷基、卤素等,例如甲苯基。术语“芳基”还包括具有两个或更多个环的多环环系,其中两个或更多个碳为两个相邻环所共有(所述环为“稠环”),其中至少一个环是芳族的且其它环例如可以是环烷基、环烯基、环炔基、芳基和/或杂环基。多环的实例包括但不限于2,3-二氢-1,4-苯并二氧杂环己二烯和2,3-二氢-1-苯并呋喃。
[0177]
本发明所述“亚烃基”为上述“烃基”脱除一个氢后的基团。
[0178]
本发明所述“亚芳基”为上述“芳香基团”脱除一个氢后的基团。
[0179]
本发明所述“亚烷基”为上述“烷基”脱除一个氢后的基团。
[0180]
本发明所述单独使用或用作后缀或前缀的“酰胺基”是指r
a-c(=o)-nh-基团,其中,r
a
选自未取代或任选被一个或多个r
b
取代的下列基团:烷基、环烷基、烯基、环烯基、炔基、环炔基、杂环基、芳基、杂芳基等;r
b
选自未取代或任选被一个或多个r
b1
取代的下列基团:卤素、羟基、巯基、硝基、氰基、烷基、烷氧基、环烷基、烯基、炔基、杂环基、芳基、杂芳基、氨基、羧基、酯基、肼基、酰基、亚磺酰基、磺酰基、磷酰基等;每一个r
b1
彼此独立地选自卤素、羟基、烷基、芳基。
[0181]
本发明所述单独使用或用作后缀或前缀的“酰肼基”是指r
a-c(=o)-nh-nh-基团,其中,r
a
的定义同前。
[0182]
本发明所述“酰胺残基”为上述“酰胺基”脱除一个氢后的基团。
[0183]
本发明所述“酰肼残基”为上述“酰肼基”脱除一个氢后的基团。
[0184]
本发明使用的术语“环烷基”意在包括具有指定数目碳原子的饱和环基。这些术语可包括稠合或桥接的多环系统。环烷基在其环结构中具有3至40个碳原子。在一个实施方案中,环烷基在其环结构中具有3、4、5或6个碳原子。例如,“c
3-6
环烷基”表示例如环丙基、环丁基、环戊基或环己基的基团。
[0185]
本发明使用的术语“环烯基”意在包括具有指定数目碳原子的含至少一个烯基的环基。这些术语可包括稠合或桥接的多环系统。环烯基在其环结构中具有3至40个碳原子。在一个实施方案中,环烯基在其环结构中具有3、4、5或6个碳原子。例如,“c
3-6
环烯基”表示例如环丙烯基、环丁烯基、环戊烯基或环己烯基的基团。
[0186]
本发明使用的术语“环炔基”意在包括具有指定数目碳原子的含至少一个炔基的环基。这些术语可包括稠合或桥接的多环系统。环炔基在其环结构中具有6至40个碳原子。在一个实施方案中,环炔基在其环结构中具有6个碳原子。例如,“c
3-6
环炔基”表示例如环丙炔基、环丁炔基、环戊炔基或环己炔基的基团。
[0187]
本发明使用的“杂芳基”指具有至少一个环杂原子(例如硫、氧或氮)的杂芳族杂
环。杂芳基包括单环系统和多环系统(例如具有2、3或4个稠环)。杂芳基的实例包括但不限于吡啶基、嘧啶基、吡嗪基、哒嗪基、三嗪基、呋喃基、喹啉基、异喹啉基、噻吩基、咪唑基、噻唑基、吲哚基、吡咯基、噁唑基、苯并呋喃基、苯并噻吩基、苯并噻唑基、异噁唑基、吡唑基、三唑基、四唑基、吲唑基、1,2,4-噻二唑基、异噻唑基、苯并噻吩基、嘌呤基、咔唑基、苯并咪唑基、苯并噁唑基、氮杂苯并噁唑基、咪唑并噻唑基、苯并[1,4]二氧杂环己烯基、苯并[1,3]二氧杂环戊烯基等。在一些实施方案中,杂芳基具有3至40个碳原子且在其它实施方案中具有3至20个碳原子。在一些实施方案中,杂芳基包含3至14个、4至14个、3至7个或5至6个成环原子。在一些实施方案中,杂芳基具有1至4个、1至3个或1至2个杂原子。在一些实施方案中,杂芳基具有1个杂原子。
[0188]
本发明使用的术语“杂环基”指包含3至20个原子的饱和、不饱和或部分饱和的单环、二环或三环,其中1、2、3、4或5个环原子选自氮、硫、氧或磷,除非另有说明,其可通过碳或氮来连接,其中-ch
2-基团任选被-c(o)-代替;其中除非另有相反说明,环氮原子或环硫原子任选被氧化以形成n-氧化物或s-氧化物或环氮原子任选被季铵化;其中环中的-nh任选被乙酰基、甲酰基、甲基或甲磺酰基取代;及环任选被一个或多个卤素取代。应该理解的是,当杂环基中s原子和o原子的总数超过1时,这些杂原子不彼此相邻。若所述杂环基为二环或三环,则至少一个环可任选为杂芳族环或芳族环,条件是至少一个环是非杂芳族的。若所述杂环基为单环,则其一定不是芳族的。杂环基的实例包括但不限于哌啶基、n-乙酰基哌啶基、n-甲基哌啶基、n-甲酰基哌嗪基、n-甲磺酰基哌嗪基、高哌嗪基、哌嗪基、氮杂环丁烷基、氧杂环丁烷基、吗啉基、四氢异喹啉基、四氢喹啉基、二氢吲哚基、四氢吡喃基、二氢-2h-吡喃基、四氢呋喃基、四氢噻喃基、四氢噻喃-1-氧化物、四氢噻喃-1,1-二氧化物、1h-吡啶-2-酮和2,5-二氧代咪唑烷基。
[0189]
本发明使用的术语“酰基”是指r
a-c(=o)-基团,其中,r
a
的定义同前。
[0190]
本发明使用的术语“亚磺酰基”是指r
a-s(=o)-基团,其中,r
a
的定义同前。
[0191]
本发明使用的术语“磺酰基”是指r
a-s(=o)
2-基团,其中,r
a
的定义同前。
[0192]
本发明使用的术语“磷酰基”是指r
c-p(=o)(r
d
)-基团,其中,r
c
和r
d
相同或不同,彼此独立地选自未取代或任选被一个或多个r
b
取代的下列基团:烷基、环烷基、烷氧基、羟基、烯基、环烯基、炔基、环炔基、杂环基、芳基、杂芳基等,r
b
的定义同前。
[0193]
本发明使用的术语“肼基”是指-nhnhr
a
基团,r
a
的定义同前。
[0194]
本发明使用的术语“胺基”指-nhr
a
基团或-n(r
a
)2基团,r
a
的定义同前。
[0195]
本发明使用的术语“氨基”指-nh2基团。
[0196]
本发明使用的术语“羧基”是指-cooh基团。
[0197]
本发明使用的术语“酯基”是指r
a-c(=o)-o-基团或r
a-o-c(=o)-基团,其中,r
a
的定义同前。
[0198]
[巯基改性高分子化合物的制备方法]
[0199]
如前所述,本发明提供一种上述巯基改性高分子化合物的制备方法,其包括以下步骤:
[0200]
1)结构上含有-cooh、-nh2、-oh中的至少一种的高分子化合物的丙烯酰化步骤,即将高分子化合物的结构上含有的-cooh、-nh2、-oh的至少一种直接或间接与如下基团连接:
[0201][0202]
r1、r2和r3的定义同前;*表示连接点;
[0203]
或者,结构上含有上述式a所示丙烯酸酯类基团、上述式b所示丙烯酰胺类基团、上述式c所示丙烯酰类基团中至少一种的高分子化合物直接作为反应原料;
[0204]
2)将步骤1)得到的高分子化合物的至少一种与多巯基化合物hs-r
4-sh反应,r4的定义同前,制备得到所述的巯基改性高分子化合物。
[0205]
在本发明的一个具体实施方式中,所述方法包括以下步骤:
[0206]
1)结构上含有-cooh、-nh2、-oh中的至少一种的高分子化合物的丙烯酰化步骤,即将高分子化合物的结构上含有的-cooh、-nh2、-oh的至少一种通过-r-基团与如下基团连接或直接与如下基团连接:
[0207][0208]
r、r1、r2和r3的定义同前,*表示连接点;
[0209]
或者,结构上含有上述式a所示丙烯酸酯类基团、上述式b所示丙烯酰胺类基团、上述式c所示丙烯酰类基团中至少一种的高分子化合物直接作为反应原料;
[0210]
2)将步骤1)得到的高分子化合物的至少一种与多巯基化合物hs-r
4-sh反应,r4的定义同前,制备得到所述的巯基改性高分子化合物。
[0211]
步骤1)中,所述丙烯酰化步骤可以是通过待改性的高分子化合物与丙烯酸酯类化合物的反应实现、也可以是通过待改性的高分子化合物与丙烯酰氯类化合物或丙烯酸酐类化合物的反应实现。
[0212]
所述丙烯酸酯类化合物可以是丙烯酸烷基酯类化合物、丙烯酸芳基酯类化合物、丙烯酸缩水多元醇酯类化合物中的一种或多种。
[0213]
所述丙烯酸缩水多元醇酯类化合物中的多元醇例如为三元醇,具体可以是甘油、丁三醇、戊三醇等。
[0214]
步骤1)中,所述丙烯酰化步骤可以是常规的反应步骤,采用现有的常规条件反应即可。通常为丙烯酰氯及其衍生物或丙烯酸酐及其衍生物与含有-oh、-nh2中的至少一种的高分子化合物反应获得。也可以为丙烯酸缩水甘油酯及其衍生物与含有-cooh、-oh、-nh2中的至少一种的高分子化合物反应获得。
[0215]
步骤1)中,所述丙烯酰化步骤可以是非常规的反应步骤,既采用非上述方法合成的含有式c结构的高分子化合物。
[0216]
步骤2)中,与多巯基化合物hs-r
4-sh反应在溶剂中进行。所述溶剂例如为水或有机溶剂,进一步可以是去离子水或二甲基甲酰胺。
[0217]
步骤2)中,与多巯基化合物hs-r
4-sh反应在低温到高温条件下进行。例如反应温度为0-80℃,进一步可以为10-70℃,例如可以在室温下反应。
[0218]
步骤2)中,与多巯基化合物hs-r
4-sh反应的反应时间为0.1-100小时。
[0219]
步骤2)中,与多巯基化合物hs-r
4-sh反应的ph范围为-1到15。例如反应ph可以为6-8,再例如为7。
[0220]
其中,步骤2)的反应产物进一步包括后处理步骤。
[0221]
其中,所述后处理步骤采用透析方法。具体的,将反应后的溶液装入透析袋(例如截留分子量2kda或以上的透析袋),用盐酸溶液(例如ph=4)透析数日(例如1-10天,还例如5天等),任选地换水(如每天换水或隔天换水)数次(例如两次或更多等),最后收集透析袋内溶液,干燥(如冷冻干燥)后得到固体或粘稠液体、即所述的巯基改性高分子化合物。
[0222]
本发明的方法中首次提出了多巯基化合物的巯基与丙烯酰类基团中的碳碳双键的迈克尔加成反应制备所述巯基改性高分子化合物,该方法不仅巯基化程度高,而且巯基化反应的条件温和(常温、水溶液中即可进行)、无污染,制备的巯基改性高分子化合物的纯度高、特别适合于进一步在医药、美容、医学等领域的使用。
[0223]
[巯基改性高分子化合物的应用]
[0224]
如上所述,本发明的巯基改性高分子化合物的巯基化程度高,适用于任何现有巯基改性高分子化合物的应用领域,具体的,其可以用于抗氧化保健品,生物医药,医疗美容整形,以及化妆品(如抗氧化类化妆品、保水补湿类化妆品中的至少一种)等领域。
[0225]
以巯基改性透明质酸为例,已知的,透明质酸(hyaluronic acid,ha)是一种线性、非支化,由双糖反复交替连接而组成的大分子酸性粘多糖聚合物(结构单元分别是β-(1,4)-n-乙酰-d-氨基葡萄糖和β-(1,3)-d-葡萄糖醛酸)。ha链长度为从约5kda到20mda之间不等,最常见的大小范围为从2mda到5mda。超过50%的透明质酸存在于皮肤、肺和肠组织中。此外,在关节滑液、软骨、脐带、血管壁等组织间质中也有存在。人体内透明质酸主要发挥润滑及缓冲作用、填充和扩散屏障、以及清除自由基等生理功能。目前市场上所用的透明质酸产品可以从动物组织(如鸡冠、眼玻璃体、脑软骨、关节液)中提取,也可由细菌(如链球菌、绿脓杆菌等)发酵而制备。近年来,随着对ha功能的深入研究,ha已广泛应用于医药领域,如用于制备药物传递系统,用于骨科疾病和眼科疾病的治疗、手术后粘连的预防及软组织修复等,该方向已成为组织工程与再生医学领域的研究热点。尽管天然ha具有极好的生物相容性和抗炎等作用,单纯ha存在较弱的机械强度以及易降解性。因此作为组织工程支架材料,ha必须经过适当的化学改性或与其他材料搭配使用,以改善这些不足,更好地发挥其生物作用,为细胞的存活和功能及组织的修复和再生提供良好的环境。
[0226]
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的保护范围。此外,应理解,在阅读了本发明所公开的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本发明所限定的保护范围之内。
[0227]
本发明中,所述1h-nmr谱图采用varian 400mhz核磁共振仪测定,测试温度25摄氏度,弛豫时间1秒钟,扫描次数为8次。具体的,取待检测物8-10毫克,溶解于750微升氘代水中,所得样品溶液测试1h-nmr谱图。
[0228]
制备例1合成丙烯酸酯修饰的透明质酸(简称ha-a1)
[0229]
在200毫升烧杯中加入1克透明质酸(购自华熙福瑞达公司,其重均分子量约为300kda),50毫升去离子水,50毫升二甲基甲酰胺,12毫升三乙胺,14毫升丙烯酸缩水甘油酯。室温搅拌至均一透明后,继续搅拌48小时。加入300毫升丙酮,产生大量白色沉淀。经离
心所得沉淀溶解于100毫升去离子水中,得到无色透明溶液。上述溶液装入透析袋(截留分子量8kda),用5升去离子水透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到921毫克白色絮状固体,即得ha-a1,收率为92.1%。
[0230]
ha-a1的结构式见图37。图37仅为示意图,表示所述透明质酸部分重复单元中的cooh被丙烯酸缩水甘油酯酯化,即其中m2/(m1+m2)表示丙烯酰化程度,m1+m2=n,n为未改性透明质酸的重复单元数。下面的制备例和实施例中的结构式的含义与制备例1的相同,就不再重复说明了。
[0231]
ha-a1的1h-nmr谱图见图37,可见位于6-6.5ppm之间的属于丙烯酸官能团的核磁峰,证明该基团成功接枝到透明质酸的结构中。
[0232]
制备例2合成丙烯酸酯修饰的透明质酸(简称ha-a2)
[0233]
在200毫升烧杯中加入1克透明质酸(购自华熙福瑞达公司,其重均分子量约为400kda),50毫升去离子水,50毫升二甲基甲酰胺,6.3克丙烯酸酐,搅拌溶解。用1摩尔每升naoh维持溶液ph=8
±
0.5,继续搅拌24小时。加入300毫升丙酮,产生大量白色沉淀。经离心所得沉淀溶解于100毫升去离子水中,得到无色透明溶液。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升去离子水透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到789毫克白色絮状固体,即得ha-a2,收率为78.9%。
[0234]
ha-a2的结构式见图38。
[0235]
ha-a2的1h-nmr谱图见图38,可见位于5.8-6.4ppm之间的属于丙烯酸官能团的核磁峰,证明该基团成功接枝到透明质酸的结构中。
[0236]
制备例3合成甲基丙烯酸酯修饰的透明质酸(简称ha-ma1)
[0237]
在200毫升烧杯中加入1克透明质酸(购自华熙福瑞达公司,其重均分子量约为400kda),50毫升去离子水,50毫升二甲基甲酰胺(sigma),12毫升三乙胺(sigma),15毫升甲基丙烯酸缩水甘油酯。室温搅拌至均一透明后,继续搅拌48小时。加入300毫升丙酮(sigma),产生大量白色沉淀。经离心所得沉淀溶解于100毫升去离子水中,得到无色溶液。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升去离子水透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到859毫克白色絮状固体,即得ha-ma1,收率为85.9%。
[0238]
ha-ma1的结构式见图39。
[0239]
ha-ma1的1h-nmr谱图见图39,可见位于5.8-6.2ppm之间的属于甲基丙烯酸官能团的核磁峰,证明该基团成功接枝到透明质酸的结构中。
[0240]
制备例4合成甲基丙烯酸酯修饰的透明质酸(简称ha-ma2)
[0241]
在200毫升烧杯中加入1克透明质酸(购自华熙福瑞达公司,其重均分子量约为400kda),100毫升去离子水,室温搅拌溶解。进一步加入7.7克甲基丙烯酸酐搅拌溶解。用1摩尔每升naoh维持溶液ph=8
±
0.5,继续搅拌24小时。加入200毫升丙酮(sigma),产生大量白色沉淀。经离心所得沉淀溶解于100毫升去离子水中,得到无色透明溶液。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升去离子水透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到846毫克白色絮状固体,即ha-ma2,收率为84.6%。
[0242]
ha-ma2的结构式见图40。
[0243]
ha-ma2的1h-nmr谱图见图40,可见位于5.8-6.2ppm之间的属于甲基丙烯酸官能团
的核磁峰,证明该基团成功接枝到透明质酸的结构中。
[0244]
制备例5合成丙烯酸酯修饰的硫酸软骨素(简称chs-a)
[0245]
在200毫升烧杯中加入1.2克硫酸软骨素(其重均分子量约为80kda),50毫升去离子水,50毫升二甲基甲酰胺,5.4克丙烯酸酐,搅拌溶解。用1摩尔每升naoh维持溶液ph=8
±
0.5,继续搅拌24小时。上述溶液装入透析袋(截留分子量3.5kda,仕必纯),用5升去离子水透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到781毫克浅黄色絮状固体,即chs-a,收率65.1%。
[0246]
chs-a的结构式见图41。
[0247]
chs-a的1h-nmr谱图见图41,可见位于6.0-6.5ppm之间的丙烯酸官能团的核磁峰,证明该基团成功接枝到硫酸软骨素的结构中。
[0248]
制备例6合成甲基丙烯酸酯修饰的硫酸软骨素(简称chs-ma)
[0249]
在200毫升烧杯中加入1.2克硫酸软骨素(其重均分子量约为90kda),50毫升去离子水,50毫升二甲基甲酰胺,进一步加入6.5克甲基丙烯酸酐搅拌溶解。用1摩尔每升naoh维持溶液ph=8
±
0.5,继续搅拌24小时。上述溶液装入透析袋(截留分子量3.5kda,仕必纯),用5升去离子水透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到776毫克浅黄色絮状固体,即chs-ma,收率64.7%。
[0250]
chs-ma的结构式见图42。
[0251]
chs-ma的1h-nmr谱图见图42,可见位于6.0-6.5ppm之间的属于甲基丙烯酸官能团的核磁峰,证明该基团成功接枝到硫酸软骨素的结构中。
[0252]
制备例7合成丙烯酸酯修饰的明胶(简称gelatin-a)
[0253]
在200毫升烧杯中加入1克明胶(强度为300blooms),50毫升去离子水,50毫升二甲基甲酰胺,进一步加入10克丙烯酸酐搅拌溶解。用1摩尔每升naoh维持溶液ph=8
±
0.5,继续搅拌24小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升去离子水透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到781毫克浅黄色絮状固体,即gelatin-a,收率78.1%。
[0254]
gelatin-a的结构简式见图43(其中的波浪线代表明胶的主链)。
[0255]
gelatin-a的1h-nmr谱图见图43,可见位于6.0-6.5ppm之间的属于丙烯酸官能团的核磁峰,证明该基团成功接枝到明胶的结构中。
[0256]
制备例8合成甲基丙烯酸酯修饰的明胶(简称gelatin-ma)
[0257]
在200毫升烧杯中加入1克明胶(强度为300blooms),50毫升去离子水,50毫升二甲基甲酰胺,进一步加入10克甲基丙烯酸酐搅拌溶解。用1摩尔每升naoh维持溶液ph=8
±
0.5,继续搅拌24小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升去离子水透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到824毫克浅黄色絮状固体,即gelatin-ma,收率82.4%。
[0258]
gelatin-ma的结构简式见图44(其中的波浪线代表明胶的主链)。
[0259]
gelatin-ma的1h-nmr谱图见图44,可见位于5.7-6.2ppm之间的属于甲基丙烯酸官能团的核磁峰,证明该基团成功接枝到明胶的结构中。
[0260]
制备例9合成丙烯酸酯修饰的乙二醇壳聚糖(简称cts-a)
[0261]
在200毫升烧杯中加入1克乙二醇壳聚糖(其重均分子量约为250kda),50毫升去离
子水,50毫升二甲基甲酰胺,8毫升三乙胺(sigma),13毫升丙烯酸缩水甘油酯。室温搅拌至均一透明后,继续搅拌48小时。上述溶液装入透析袋(截留分子量3.5kda,仕必纯),用5升去离子水透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到694毫克浅黄色絮状固体,即cts-a,收率69.4%。
[0262]
cts-a结构式见图45。
[0263]
cts-a的1h-nmr谱图见图45,可见位于5.8-6.4ppm之间的属于丙烯酸官能团的核磁峰,证明该基团成功接枝到乙二醇壳聚糖的结构中。
[0264]
制备例10合成甲基丙烯酸酯修饰的乙二醇壳聚糖(简称cts-ma)
[0265]
在200毫升烧杯中加入1克乙二醇壳聚糖(其重均分子量约为200kda),50毫升去离子水,50毫升二甲基甲酰胺,8毫升三乙胺(sigma),13毫升甲基丙烯酸缩水甘油酯。室温搅拌至均一透明后,继续搅拌48小时。上述溶液装入透析袋(截留分子量3.5kda,仕必纯),用5升去离子水透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到726毫克浅黄色絮状固体,即cts-ma,收率72.6%。
[0266]
cts-ma的结构式见图46。
[0267]
cts-ma的1h-nmr谱图见图46,可见位于5.7-6.2ppm之间的属于甲基丙烯酸官能团的核磁峰,证明该基团成功接枝到乙二醇壳聚糖的结构中。
[0268]
制备例11合成丙烯酸酯修饰的聚甲基丙烯酸羟乙酯(简称phema-a)
[0269]
在200毫升烧杯中加入2克聚甲基丙烯酸羟乙酯(mv=20kda,购自sigma公司),50毫升去离子水,50毫升二甲基甲酰胺进一步加入16.5克丙烯酸酐搅拌溶解。用1摩尔每升naoh维持溶液ph=8
±
0.5,继续搅拌24小时。上述溶液装入透析袋(截留分子量2kda,仕必纯),用5升去离子水透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到1.42克白色固体,即phema-a,收率71.0%。
[0270]
phema-a的结构式见图47。
[0271]
phema-a的1h-nmr谱图见图47,可见位于5.9-6.4ppm之间的属于丙烯酸官能团的核磁峰,证明该基团成功接枝到聚甲基丙烯酸羟乙酯的结构中。
[0272]
制备例12合成甲基丙烯酸酯修饰的聚甲基丙烯酸羟乙酯(简称phema-ma)
[0273]
在200毫升烧杯中加入2克聚甲基丙烯酸羟乙酯(mv=20kda,购自sigma公司),50毫升去离子水,50毫升二甲基甲酰胺,进一步加入16.8克甲基丙烯酸酐搅拌溶解。用1摩尔每升naoh维持溶液ph=8
±
0.5,继续搅拌24小时。上述溶液装入透析袋(截留分子量2kda,仕必纯),用5升去离子水透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到1.48克白色固体,即phema-ma,收率74.0%。
[0274]
phema-ma的结构式见图48。
[0275]
phema-ma的1h-nmr谱图见图48,可见位于5.7-6.3ppm之间的属于甲基丙烯酸官能团的核磁峰,证明该基团成功接枝到聚甲基丙烯酸羟乙酯的结构中。
[0276]
制备例13合成丙烯酸酯修饰的聚乙烯醇(简称pva-a)
[0277]
在200毫升烧杯中加入2克聚乙烯醇(mw=61kda,购自sigma公司),50毫升去离子水,50毫升二甲基甲酰胺,进一步加入13克丙烯酸酐搅拌溶解。用1摩尔每升naoh维持溶液ph=8
±
0.5,继续搅拌24小时。上述溶液装入透析袋(截留分子量3.5kda,仕必纯),用5升去离子水透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到1.57克白色固
体,即pva-a,收率78.5%。
[0278]
pva-a的结构式见图49。
[0279]
pva-a的1h-nmr谱图见图49,可见位于6.0-6.5ppm之间的属于丙烯酸官能团的核磁峰,证明该基团成功接枝到聚乙烯醇的结构中。
[0280]
制备例14合成甲基丙烯酸酯修饰的聚乙烯醇(简称pva-ma)
[0281]
在200毫升烧杯中加入2克聚乙烯醇(mw=61kda,购自sigma公司),50毫升去离子水,50毫升二甲基甲酰胺,进一步加入13.4克甲基丙烯酸酐搅拌溶解。用1摩尔每升naoh维持溶液ph=8
±
0.5,继续搅拌24小时。上述溶液装入透析袋(截留分子量3.5kda,仕必纯),用5升去离子水透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到1.51克白色固体,即pva-ma,收率75.5%。
[0282]
pva-ma的结构式见图50。
[0283]
pva-ma的1h-nmr谱图见图50,可见位于5.7-6.3ppm之间的属于甲基丙烯酸官能团的核磁峰,证明该基团成功接枝到聚乙烯醇的结构中。
[0284]
实施例1合成巯基-丙烯酸酯修饰的透明质酸(简称ha-a1-sh1)
[0285]
在200毫升烧杯中加入1克按制备例1的方法制备的ha-a1,0.3克二硫苏糖醇(购自vwr公司),100毫升去离子水,室温搅拌溶解,得到透明溶液。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到842毫克白色絮状固体,即得ha-a1-sh1,收率为84.2%。
[0286]
ha-a1-sh1的反应方程式如图9所示,其结构式见图9和图51。
[0287]
ha-a1-sh1的1h-nmr谱图见图51,可见位于2.3-2.8ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到透明质酸的结构中。
[0288]
实施例2合成巯基-丙烯酸酯修饰的透明质酸(简称ha-a2-sh1)
[0289]
在200毫升烧杯中加入1克按制备例2的方法制备的ha-a2,0.3克二硫苏糖醇(vwr),100毫升去离子水,室温搅拌溶解,得到透明溶液。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到827毫克白色絮状固体,即得ha-a2-sh1,收率为82.7%。
[0290]
ha-a2-sh1的反应方程式如图10所示,其结构式见图10和图52。
[0291]
ha-a2-sh1的1h-nmr谱图见图52,可见位于2.6-2.9ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到透明质酸的结构中。
[0292]
实施例3合成巯基-甲基丙烯酸酯修饰的透明质酸(简称ha-ma1-sh1)
[0293]
在200毫升烧杯中加入1克按制备例3的方法制备的ha-ma1,0.3克二硫苏糖醇(vwr),100毫升去离子水,室温搅拌溶解。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到白色絮状固体约854毫克。即得ha-ma1-sh1,收率为85.4%。
[0294]
ha-ma1-sh1的反应方程式如图11所示,其结构式见图11和图53。
[0295]
ha-ma1-sh1的1h-nmr谱图见图53,可见位于2.6-3.0ppm之间的属于巯基侧链的核
磁峰,证明巯基成功接枝到透明质酸的结构中。
[0296]
实施例4合成巯基-甲基丙烯酸酯修饰的透明质酸(简称ha-ma2-sh1)
[0297]
在200毫升烧杯中加入1克按制备例4的方法制备的ha-ma2,0.3克二硫苏糖醇(vwr),100毫升去离子水,室温搅拌溶解。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到白色絮状固体约833毫克。即得ha-ma2-sh1,收率为83.3%。
[0298]
ha-ma2-sh1的反应方程式如图12所示,其结构式见图12和图54。
[0299]
ha-ma2-sh1的1h-nmr谱图见图54,可见位于2.6-3.0ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到透明质酸的结构中。
[0300]
实施例5合成巯基-丙烯酸酯修饰的硫酸软骨素(简称chs-a-sh1)
[0301]
在200毫升烧杯中加入1克按制备例5的方法制备的chs-a,0.25克二硫苏糖醇(vwr),100毫升去离子水,室温搅拌溶解。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量3.5kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到浅黄色絮状固体629毫克,即得chs-a-sh1,收率为62.9%。
[0302]
chs-a-sh1的反应方程式如图13所示,其结构式见图13和图55。
[0303]
chs-a-sh1的1h-nmr谱图见图55,可见位于2.6-3.0ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到硫酸软骨素的结构中。
[0304]
实施例6合成巯基-甲基丙烯酸酯修饰的硫酸软骨素(简称chs-ma-sh1)
[0305]
在200毫升烧杯中加入1克按制备例6的方法制备的chs-ma,0.25克二硫苏糖醇(vwr),100毫升去离子水,室温搅拌溶解。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量3.5kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到浅黄色絮状固体642毫克,即得chs-ma-sh1,收率为64.2%。
[0306]
chs-ma-sh1反应方程式如图14所示,其结构式见图14和图56。
[0307]
chs-ma-sh1的1h-nmr谱图见图56,可见位于2.6-3.0ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到硫酸软骨素的结构中。
[0308]
实施例7合成巯基-丙烯酸酯修饰的明胶(简称gelatin-a-sh1)
[0309]
在200毫升烧杯中加入1克按制备例7的方法制备的gelatin-a,0.19克二硫苏糖醇(vwr),100毫升去离子水,室温搅拌溶解。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到浅黄色絮状固体763毫克,即得gelatin-a-sh1,收率为76.3%。
[0310]
gelatin-a-sh1的反应方程式如图15所示,其结构式见图15和图57。
[0311]
gelatin-a-sh1的1h-nmr谱图见图57,可见位于2.6-2.8ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到明胶的结构中。
[0312]
实施例8合成巯基-甲基丙烯酸酯修饰的明胶(简称gelatin-ma-sh1)
[0313]
在200毫升烧杯中加入1克按制备例8的方法制备的gelatin-ma,0.19克二硫苏糖
醇(vwr),100毫升去离子水,室温搅拌溶解。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到浅黄色絮状固体787毫克,即得gelatin-ma-sh1,收率为78.7%
[0314]
gelatin-ma-sh1的反应方程式如图16所示,其结构式见图16和图58。
[0315]
gelatin-ma-sh1的1h-nmr谱图见图58,可见位于2.6-2.7ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到明胶的结构中。
[0316]
实施例9合成巯基-丙烯酸酯修饰的乙二醇壳聚糖(简称cts-a-sh1)
[0317]
在200毫升烧杯中加入1克按制备例9的方法制备的cts-a,0.25克二硫苏糖醇(vwr),100毫升去离子水,室温搅拌溶解。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量3.5kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到浅黄色絮状固体602毫克,即得cts-a-sh1,收率为60.2%。
[0318]
cts-a-sh1的反应方程式如图17所示,其结构式见图17和图59。
[0319]
cts-a-sh1的1h-nmr谱图见图59,可见位于2.6-3.0ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到乙二醇壳聚糖的结构中。
[0320]
实施例10合成巯基-甲基丙烯酸酯修饰的壳聚糖(简称cts-ma-sh1)
[0321]
在200毫升烧杯中加入1克按制备例10的方法制备的cts-ma,0.25克二硫苏糖醇(vwr),100毫升去离子水,室温搅拌溶解。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量3.5kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到白色絮状固体643毫克,即得cts-ma-sh1,收率为64.3%
[0322]
cts-ma-sh1的反应方程式如图18所示,其结构式见图18和图60。
[0323]
cts-ma-sh1的1h-nmr谱图见图60,可见位于2.5-2.9ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到壳聚糖的结构中。
[0324]
实施例11合成巯基-丙烯酸酯修饰的聚甲基丙烯酸羟乙酯(简称phema-a-sh1)
[0325]
在200毫升烧杯中加入2克按制备例11的方法制备的phema-a,0.42克二硫苏糖醇(vwr),50毫升去离子水,50毫升二甲基甲酰胺,室温搅拌溶解。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量2kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到白色固体1.67克,即得phema-a-sh1,收率为83.5%。
[0326]
phema-a-sh1的反应方程式如图19所示,其结构式见图19和图61。
[0327]
phema-a-sh1的1h-nmr谱图见图61,可见位于2.6-2.9ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到聚甲基丙烯酸羟乙酯的结构中。
[0328]
实施例12合成巯基-甲基丙烯酸酯修饰的聚甲基丙烯酸羟乙酯(简称phema-ma-sh1)
[0329]
在200毫升烧杯中加入2克按制备例12的方法制备的phema-ma,0.41克二硫苏糖醇(vwr),50毫升去离子水,50毫升二甲基甲酰胺,室温搅拌溶解。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量2kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天
换水两次。最后收集透析袋内溶液,经冷冻干燥后得到白色固体1.62克,即得phema-ma-sh1,收率为81%。
[0330]
phema-ma-sh1的反应方程式如图20所示,其结构式见图20和图62。
[0331]
phema-ma-sh1的1h-nmr谱图见图62,可见位于2.6-3.0ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到聚甲基丙烯酸羟乙酯的结构中。
[0332]
实施例13合成巯基修饰的超支化peg聚合物(简称hb-peg-sh1)
[0333]
在200毫升烧杯中加入5克超支化peg即hb-peg(mw=20kda,购自blafar ltd),0.86克二硫苏糖醇(vwr),100毫升去离子水,室温搅拌溶解。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量2kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到无色粘稠液体3.84克,即得hb-peg-sh1,收率为76.8%。
[0334]
hb-peg-sh1的反应方程式如图21所示,其结构式见图21和图63。
[0335]
hb-peg-sh1的1h-nmr谱图见图63,可见位于2.5-2.6ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到超支化peg聚合物的结构中。
[0336]
实施例14合成巯基-丙烯酸酯修饰的聚乙烯醇(简称pva-a-sh1)
[0337]
在200毫升烧杯中加入1克按制备例13的方法制备的pva-a,100毫升去离子水,溶液加热搅拌至pva-a完全溶解。随后溶液中加入0.47克二硫苏糖醇(vwr),室温搅拌溶解。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到白色固体737毫克,即得pva-a-sh1,收率为73.7%。
[0338]
pva-a-sh1的反应方程式如图22所示,其结构式见图22和图64。
[0339]
pva-a-sh1的1h-nmr谱图见图64,可见位于2.6-3.0ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到聚乙烯醇的结构中。
[0340]
实施例15合成巯基-甲基丙烯酸酯修饰的聚乙烯醇(简称pva-ma-sh1)
[0341]
在200毫升烧杯中加入1克按制备例14的方法制备的pva-ma,100毫升去离子水,溶液加热搅拌至pva-ma完全溶解。随后溶液中加入0.47克二硫苏糖醇(vwr),室温搅拌溶解。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到白色固体718毫克,即得pva-ma-sh1,收率为71.8%。
[0342]
pva-ma-sh1的反应方程式如图23所示,其结构式见图23和图65。
[0343]
pva-ma-sh1的1h-nmr谱图见图65,可见位于2.5-3.0ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到聚乙烯醇的结构中。
[0344]
实施例16合成巯基-丙烯酸酯修饰的透明质酸(简称ha-a1-sh2)
[0345]
在200毫升烧杯中加入1克按制备例1的方法制备的ha-a1,0.42克1,4-丁二硫醇(购自sigma公司),100毫升去离子水,室温搅拌溶解,得到透明溶液。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到852毫克白色絮状固体,即得ha-a1-sh2,收率为85.2%。
[0346]
ha-a1-sh2的反应方程式如图24所示,其结构式见图24和图66。
[0347]
ha-a1-sh2的1h-nmr谱图见图66,可见位于1.6-1.9ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到透明质酸的结构中。
[0348]
实施例17合成巯基-丙烯酸酯修饰的透明质酸(简称ha-a1-sh3)
[0349]
在200毫升烧杯中加入1克按制备例1的方法制备的ha-a1,0.43克2-氨基-1,4-丁二硫醇盐酸盐(购自sigma公司),100毫升去离子水,室温搅拌溶解,得到透明溶液。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到843毫克白色絮状固体,即得ha-a1-sh3,收率为84.3%。
[0350]
ha-a1-sh3的反应方程式如图25所示,其结构式见图25和图67。
[0351]
ha-a1-sh3的1h-nmr谱图见图67,可见位于3.0-3.2ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到透明质酸的结构中。
[0352]
实施例18合成巯基-丙烯酸酯修饰的透明质酸(简称ha-a2-sh2)
[0353]
在200毫升烧杯中加入1克按制备例2的方法制备的ha-a2,0.42克1,4-丁二硫醇(购自sigma公司),100毫升去离子水,室温搅拌溶解,得到透明溶液。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到827毫克白色絮状固体,即得ha-a2-sh2,收率为82.7%。
[0354]
ha-a2-sh2的反应方程式如图26所示,其结构式见图26和图68。
[0355]
ha-a2-sh2的1h-nmr谱图见图68,可见位于1.6-1.9ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到透明质酸的结构中。
[0356]
实施例19合成巯基-丙烯酸酯修饰的透明质酸(简称ha-a2-sh3)
[0357]
在200毫升烧杯中加入1克按制备例2的方法制备的ha-a2,0.43克2-氨基-1,4-丁二硫醇盐酸盐(sigma),100毫升去离子水,室温搅拌溶解,得到透明溶液。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到833毫克白色絮状固体,即得ha-a2-sh3,收率为83.3%。
[0358]
ha-a2-sh3的反应方程式如图27所示,其结构式见图27和图69。
[0359]
ha-a2-sh3的1h-nmr谱图见图69,可见位于3.0-3.2ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到透明质酸的结构中。
[0360]
实施例20合成巯基-丙烯酸酯修饰的透明质酸(简称ha-a2-sh4)
[0361]
在200毫升烧杯中加入1克按制备例2的方法制备的ha-a2,0.38克1,3-丙二硫醇(购自sigma公司),100毫升去离子水,室温搅拌溶解,得到透明溶液。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到814毫克白色絮状固体,即得ha-a2-sh4,收率为81.4%。
[0362]
ha-a2-sh4的反应方程式如图28所示,其结构式见图28和图70。
[0363]
ha-a2-sh4的1h-nmr谱图见图70,可见位于2.5-2.8ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到透明质酸的结构中。
[0364]
实施例21合成巯基-丙烯酸酯修饰的透明质酸(简称ha-a2-sh5)
[0365]
在200毫升烧杯中加入1克按制备例2的方法制备的ha-a2,0.52克1,3-苯二硫酚(购自sigma公司),100毫升去离子水,室温搅拌溶解,得到透明溶液。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到836毫克白色絮状固体,即得ha-a2-sh5,收率为83.6%。
[0366]
ha-a2-sh5的反应方程式如图29所示,其结构式见图29和图71。
[0367]
ha-a2-sh5的1h-nmr谱图见图71,可见位于6.9-7.4ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到透明质酸的结构中。
[0368]
实施例22合成巯基-丙烯酸酯修饰的透明质酸(简称ha-a2-sh6)
[0369]
在200毫升烧杯中加入1克按制备例2的方法制备的ha-a2,0.52克1,4-苯二硫酚(购自sigma公司),100毫升去离子水,室温搅拌溶解,得到透明溶液。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到831毫克白色絮状固体,即得ha-a2-sh6,收率为83.1%。
[0370]
ha-a2-sh6的反应方程式如图30所示,其结构式见图30和图72。
[0371]
ha-a2-sh6的1h-nmr谱图见图72,可见位于6.8-7.0ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到透明质酸的结构中。
[0372]
实施例23合成巯基-丙烯酸酯修饰的透明质酸(简称ha-a2-sh7)
[0373]
在200毫升烧杯中加入1克按制备例2的方法制备的ha-a2,0.96克巯基聚乙二醇(购自sigma公司),100毫升去离子水,室温搅拌溶解,得到透明溶液。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到894毫克白色絮状固体,即得ha-a2-sh7,收率为89.4%。
[0374]
ha-a2-sh7的反应方程式如图31所示,其结构式见图31和图73。
[0375]
ha-a2-sh7的1h-nmr谱图见图73,可见位于3.6ppm的属于巯基侧链的核磁峰,证明巯基成功接枝到透明质酸的结构中。
[0376]
实施例24合成巯基-丙烯酸酯修饰的透明质酸(简称ha-a2-sh8)
[0377]
在200毫升烧杯中加入1克按制备例2的方法制备的ha-a2,0.74克三羟甲基丙烷基-三(3-巯基丙酸酯)(购自sigma公司),50毫升去离子水和50毫升二甲基甲酰胺,室温搅拌溶解,得到透明溶液。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到785毫克白色絮状固体,即得ha-a2-sh8,收率为78.5%。
[0378]
ha-a2-sh8的反应方程式如图32所示,其结构式见图32和图74。
[0379]
ha-a2-sh8的1h-nmr谱图见图74,可见位于0.8-1.0ppm、1.5ppm、2.6-2.9ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到透明质酸的结构中。
[0380]
实施例25合成巯基-甲基丙烯酸酯修饰的透明质酸(简称ha-ma1-sh5)
[0381]
在200毫升烧杯中加入1克按制备例3的方法制备的ha-ma1,0.50克1,3-苯二硫酚(购自sigma公司),100毫升去离子水,室温搅拌溶解,得到透明溶液。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5
天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到828毫克白色絮状固体,即得ha-ma1-sh5,收率为82.8%。
[0382]
ha-ma1-sh5的反应方程式如图33所示,其结构式见图33和图75。
[0383]
ha-ma1-sh5的1h-nmr谱图见图75,可见位于6.9-7.4ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到透明质酸的结构中。
[0384]
实施例26合成巯基-甲基丙烯酸酯修饰的透明质酸(简称ha-ma1-sh6)
[0385]
在200毫升烧杯中加入1克按制备例3的方法制备的ha-ma1,0.50克1,4-苯二硫酚(购自sigma公司),100毫升去离子水,室温搅拌溶解,得到透明溶液。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到833毫克白色絮状固体,即得ha-ma1-sh6,收率为83.3%。
[0386]
ha-ma1-sh6的反应方程式如图34所示,其结构式见图34和图76。
[0387]
ha-ma1-sh6的1h-nmr谱图见图76,可见位于6.9-7.0ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到透明质酸的结构中。
[0388]
实施例27合成巯基-甲基丙烯酸酯修饰的透明质酸(简称ha-ma2-sh7)
[0389]
在200毫升烧杯中加入1克按制备例4的方法制备的ha-ma2,0.92克巯基聚乙二醇(购自sigma公司),100毫升去离子水,室温搅拌溶解,得到透明溶液。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到876毫克白色絮状固体,即得ha-ma2-sh7,收率为87.6%。
[0390]
ha-ma2-sh7的反应方程式如图35所示,其结构式见图35和图77。
[0391]
ha-ma2-sh7的1h-nmr谱图见图77,可见位于3.6ppm的属于巯基侧链的核磁峰,证明巯基成功接枝到透明质酸的结构中。
[0392]
实施例28合成巯基-甲基丙烯酸酯2修饰的透明质酸(简称ha-ma2-sh8)
[0393]
在200毫升烧杯中加入1克按制备例4的方法制备的ha-ma2,0.68克三羟甲基丙烷基-三(3-巯基丙酸酯)(购自sigma公司),50毫升去离子水和50毫升二甲基甲酰胺,室温搅拌溶解,得到透明溶液。所得透明溶液继续搅拌12小时。上述溶液装入透析袋(截留分子量8kda,仕必纯),用5升ph=4的盐酸溶液透析5天,每天换水两次。最后收集透析袋内溶液,经冷冻干燥后得到825毫克白色絮状固体,即得ha-ma2-sh8,收率为82.5%。
[0394]
ha-ma2-sh8的反应方程式如图36所示,其结构式见图36和图78。
[0395]
ha-ma2-sh8的1h-nmr谱图见图78,可见位于0.8-1.0ppm、1.5ppm、2.6-2.9ppm之间的属于巯基侧链的核磁峰,证明巯基成功接枝到透明质酸的结构中。
[0396]
实施例29ellman法检测巯基改性高分子化合物的巯基含量
[0397]
准备过程:
[0398]
1.配制测试缓冲溶液:0.1摩尔每升na2hpo4(含1mm edta,使用浓盐酸调节缓冲液ph=8.0)。
[0399]
2.配制标准品工作液:30毫摩尔每升半胱氨酸溶液。
[0400]
3.配制ellman试剂母液:0.1摩尔每升ellman试剂溶液。
[0401]
4.标准品溶液配制:
[0402]
巯基摩尔浓度0mmol/l0.5mmol/l1.0mmol/l1.5mmol/l2.0mmol/l标准品工作液(μl)0481216缓冲液(μl)240236232228224总体积(μl)240240240240240
[0403]
5.产品测试样:取巯基改性高分子化合物样品适量溶解于缓冲溶液中配制成1mg/ml的待测溶液(每个样品三组平行样)。
[0404]
测试过程:
[0405]
1.半胱氨酸标准品溶液按上述步骤4配制于0.5ml离心管中。
[0406]
2.另取1.5ml离心管,将50μl ellman检测液加入1ml缓冲溶液中得到检测液。
[0407]
3.取240μl标准品溶液/测试样溶液分别混溶于测试过程步骤2中的ellman检测液中室温反应15min。
[0408]
4.15min后用孔板仪检测412nm处吸光值。
[0409]
5.产品中的硫醇含量可以根据所得标准品溶液的吸光度/浓度标准曲线计算求出。
[0410]
巯基含量检测标准曲线见图79,巯基含量检测结果见表1。
[0411]
实施例30巯基修饰高分子化合物的动力粘度检测
[0412]
取500毫克巯基修饰高分子化合物溶解于50毫升去离子水中得到浓度为1%w/v溶液。在25℃条件下使用旋转粘度计对所得溶液进行测试得到动力粘度,结果见表1(phema-a-sh1与phema-ma-sh1溶解后粘度过大,呈现凝胶状,因此未检测动力粘度)。
[0413]
表1巯基修饰高分子化合物的巯基含量及动力粘度检测结果
[0414]
[0415][0416]
实施例31gpc测定巯基修饰高分子化合物分子量及其分布
[0417]
gpc流动相为0.05m硫酸钠溶液,流速1ml/min,柱温30℃,以标准聚乙二醇聚合物作为标准曲线。
[0418]
取巯基修饰高分子化合物5毫克,溶解于1毫升0.05m硫酸钠溶液中,使用0.22微米滤头过滤后进行gpc测试,分子量及分布结果见表2(phema-a-sh1与phema-ma-sh1溶解后粘度过大,无法通过滤头,因此未检测分子量数据)。
[0419]
表2巯基修饰高分子化合物分子量和分子量分布的检测结果
[0420][0421][0422]
实施例32巯基修饰高分子化合物的保水性能测试表3:保水率指标对比表
[0423][0424][0425]
在预先称量瓶重的20毫升玻璃瓶中加入50毫克巯基修饰高分子化合物,加入5毫升去离子水溶解中得到1%浓度的溶液,由质量差减法得到溶液质量m0。将玻璃瓶置于37℃摇床内,每隔一定时间称量溶液质量得到m
t
。巯基修饰高分子化合物的保水能力可根据下式计算:
[0426]
保水百分比(%)=m
t
/m0×
100%。
[0427]
测试结果列于表3中。
[0428]
实施例33dpph自由基捕获法检测巯基修饰高分子化合物的抗氧化性
[0429]
精确配制25微摩尔每升1,1-二苯基-2-三硝基苯肼(tnbs)的绝对乙醇溶液作为工作液。取一定量工作液经乙醇稀释后得到一系列标准品溶液(0,5,10,15,20,25微摩尔每升)。
[0430]
精确配制巯基取代高分子化合物的pbs溶液得到一系列浓度为0.1mg/ml的测试样溶液。
[0431]
取tnbs工作液90微升与测试样溶液10微升混合均匀,室温避光保存30分钟后检测tnbs标准品溶液以及测试样品混合液在517nm处的吸光度,根据所得标准曲线计算得到测试样品中剩余dpph的浓度,测试样品自由基捕获能力(%)根据下式计算得出:
[0432]
自由基捕获能力(%)=(1-(c
样品
/c
dpph
))
×
100%。
[0433]
dpph标准曲线见图80。自由基捕获能力见图81。
[0434]
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1