一种4-氨基-5-咪唑甲酰胺的工业化生产方法与流程
本发明属于有机合成技术领域,具体涉及一种4-氨基-5-咪唑甲酰胺的工业化生产方法。
背景技术:
众所周知,替莫唑胺(temozolomide,简称tmz)是一种治疗脑胶质瘤的药物,该药物相比其它类型的抗肿瘤药物,有着很多自身独特的优势:如具有较宽的抗肿瘤谱、能够口服且吸收效果好、易于透过脑血屏障、耐受性好、骨髓抑制作用小、且与其它药物一起服用时不会产生叠加毒性,安全性好。更为神奇的是替莫唑胺药物除对脑胶质瘤有很好的疗效外,还对淋巴瘤、实体瘤、白血病以及黑色素瘤也有很好的治疗效果{口服抗肿瘤新药-替莫唑胺[j].医药肿瘤学:国外分册,1999,26(1):292-293.}。替莫唑胺药物是由英国schering-plough公司率先研制,于1997年在欧洲上市{抗肿瘤药替莫唑胺的合成[j].精细化工中间体,2004,34(5):27-28.},既欧洲上市以后,1999年8月11日经fda批准在美国获批上市,同时该公司已经向包括加拿大、澳大利亚、新西兰在内的16个国家提出了该药物的上市申请[替莫唑胺的合成[j].山东医药工业,2003,22(4):1.]。新型抗肿瘤药替莫唑胺的上市,给众多的脑肿瘤患者带来了福音,解除了他们的痛苦,也延长了他们的寿命,提高了他们的生活质量。
4-氨基-5-咪唑甲酰胺做为替莫唑胺药物合成的重要中间体,其合成及产业化备受广大研究者关注。目前合成4-氨基-5-咪唑甲酰胺的主要有两条合成路线:合成路线1是以氰化物为原料进行合成[如下合成路线1,wo2014013151;wo2004035529],该方法由于均用到剧毒品氰化物(氰化钠、氢氰酸、氰基丙醇),工业化生产存在着很大的局限性,因为只有具有氰化物使用资质的企业才能够用此方法进行工业化生产,绝大多数普通化工生产企业因不具备氰化物使用资质,故不能使用该合成方法进行工业化生产;另外一方面,即使是具备氰化物使用资质的化工生产企业,由于氰化物为剧毒品,生产过程中人员管理以及生产过程的废水处理,需进行非常严格的管控,不正确或者不严谨的管控措施,都将会给人员或者环境带来致命的影响。
合成路线2是以氰基乙酸乙酯为起始原料进行合成[如下合成方法2,wo2010140168;journalofthechemicalsociety,1440-1442,1949],该合成方法虽然改用了低毒起始物料氰基乙酸乙酯,该物料的大规模使用不会对人员和环境带来伤害,但方法2在合成过程中,特别是第三步反应,需用到大量的氨甲醇溶液,一方面氨甲醇大规模使用过程中气味很大,另一方面由于氨甲醇的大规模使用,必将产生大量的含氨氮的废水,环保压力非常之大,在当前严峻的环保形势下,方法2也难以实现工业化生产。
除文献报道的上述两种主要的合成方法外,还有一些其它合成方法的报道{tetrahedronletters,25(49),5701-5704,1984;canadianjournalofchemistry,59(9),1347-1356,1981;agriculturalandbiologicalchemistry,41(12),2331-2334,1977;journaloftheamericanchemicalsociety,82,3144-3146,1960;journaloforganicchemistry,24,256-257,1959;journalofbiologicalchemistry,181,89-93,1949},但这些方法,均需从很复杂的中间体原料来开始合成,其中大部分上述复杂的中间体原料,几乎都没有大规模供应,且这些复杂的中间体原料,价格普遍都很高,导致合成至4-氨基-5-咪唑甲酰胺,无法产生经济效益。
综上所述,现有的4-氨基-5-咪唑甲酰胺的合成方法均不能满足工业化生产的需要。
技术实现要素:
为了解决上述问题,本发明公开了一种4-氨基-5-咪唑甲酰胺的工业化生产方法。
一种4-氨基-5-咪唑甲酰胺的工业化生产方法,包括以下步骤:
s1:二氨基马来腈与甲酰胺在三氯氧磷作用下反应生成中间体1;
s2:中间体1在碱性条件下发生关环反应,生成4-氨基-5-咪唑甲酰胺;
其合成路线如下:
优选地,所述s1具体包括以下步骤:
惰性气体保护下,将二氨基马来腈与甲酰胺溶于thf,在0~5℃加入三氯氧磷,之后控温5~35℃反应,制得反应液,反应液后处理,制得中间体1。
优选地,所述二氨基马来腈:甲酰胺摩尔比为1:1.2~1.5,二氨基马来腈:三氯氧磷摩尔比为1:1.2~1.5;二氨基马来腈:四氢呋喃质量比为1:4.4~8。
优选地,s1控温反应时间为3h。
优选地,所述反应液后处理包括以下步骤:反应液加入甲醇搅拌,之后用水淬灭,调节ph=8~8.5,用乙酸乙酯萃取,有机相用无水硫酸镁干燥,过滤,浓缩滤液,浓缩物加入石油醚打浆,过滤,滤饼干燥即可。
优选地,所述s2具体包括以下步骤:
惰性气体保护下,将s1中间1与水和碱混合,控温95~100℃反应,制得混合液,混合液后处理,制得4-氨基-5-咪唑甲酰胺。
优选地,所述中间体1:碱的摩尔比为1:3.3~3.4,所述中间体1与水的质量比为1:5。
优选地,所述碱为氢氧化钠。
优选地,s2控温反应的时间为3h。
优选地,所述混合液后处理包括以下步骤:混合液在0~5℃用盐酸调节ph至6.5~7,过滤,滤饼水洗、干燥,用水重结晶,无水乙醇洗涤,干燥即可。
与现有技术相比,本发明的有益效果是:
(1)本发明以二氨基马来腈为原料经两步反应合成4-氨基-5-咪唑甲酰胺,是一种全新的合成方法,国内外文献中未见报道,就合成路线绿色环保而言,本发明相比背景技术中剧毒的氰化物方法(氰化钠、氢氰酸、氰基丙醇)或者氨甲醇的合成方法,本发明所用的原材料简单,低毒,不会产生含氰根(剧毒)或含氮的废水产生,仅只会产生少量的含磷的废水,本发明的合成技术对环境相对友好;
(2)就反应所需设备和工厂所需资质而言,本发明相比背景技术中剧毒的氰化物方法(氰化钠、氢氰酸、氰基丙醇)或者氨甲醇的合成方法,本发明常规反应设备即可满足工业化生产的要求,工厂也不需要特殊的资质;
(3)就最终产品品质而言,本发明相比背景技术中剧毒的氰化物方法(氰化钠、氢氰酸、氰基丙醇)或者氨甲醇的合成方法,能得到纯度99.8%以上的目标产物,且品质稳定,重复性好;
(4)就综合成本而言,本发明相比背景技术中剧毒的氰化物方法(氰化钠、氢氰酸、氰基丙醇)或者氨甲醇的合成方法,由于合成路线所用的原料简单,价格低廉,且合成总收率能到58.81%,故单位产品的原材料成本较低;由于合成路线仅为两步反应,且每步反应的反应时间及后处理时间均较短,后处理简单,故单位产品的制造成本较低;由于合成路线所用物料相对环保,不会产生大量的废水、废固及废气,故单位产品的环保成本低,综合考虑该产品的单位原材料成本、单位制造成本、单位环保成本,故该产品的综合成本较背景技术中剧毒的氰化物方法(氰化钠、氢氰酸、氰基丙醇)或者氨甲醇的合成方法均有着极为明显的优势。
附图说明
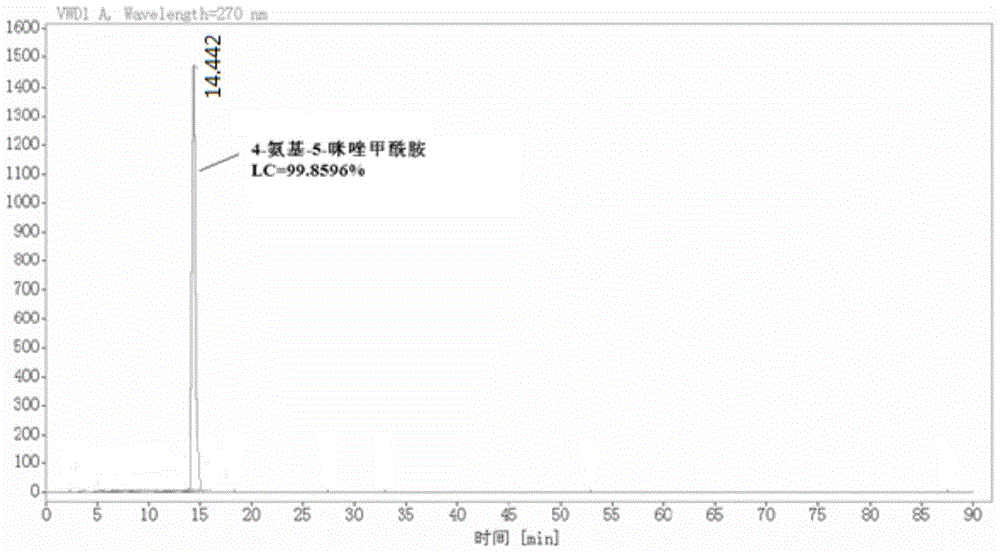
图1为实施例1制得的4-氨基-5-咪唑甲酰胺的lc图谱;
图2为实施例1制得的4-氨基-5-咪唑甲酰胺的核磁氢谱图。
具体实施方式
为了使本领域技术人员更好地理解本发明的技术方案能予以实施,下面结合具体实施例和附图对本发明作进一步说明,但所举实施例不作为对本发明的限定。
下述实施例中所述的实验方法和检测方法,如无特殊说明,均为常规方法;所述的原料和试剂,如无特殊说明,均为市售。
一种4-氨基-5-咪唑甲酰胺的工业化生产方法,包括以下步骤:
s1:二氨基马来腈与甲酰胺,在三氯氧磷作用下反应生成中间体1;
s2:s1中间体1在碱性条件下发生关环反应,生成4-氨基-5-咪唑甲酰胺;
其合成路线如下:
实施例1
一种4-氨基-5-咪唑甲酰胺的工业化生产方法,具体包括以下步骤:
步骤1,中间体1的合成:
氩气保护条件下,向装有温度计、滴加漏斗和机械搅拌的5l的三口瓶中依次加入thf1620ml、二氨基马来腈324g,甲酰胺162g(1.2eq.),低温浴槽降温至5℃,开始滴加三氯氧磷550.8g(1.2eq.),滴毕后保温5℃反应2h,开始每隔1h取样,当二氨基马来腈lc<0.3%时,停止反应。
后处理:控温20℃,向反应液中滴加甲醇345.6g,滴加完毕控温20℃反应2h后,将体系缓慢倒入水4.2l中淬灭,淬灭完保温20℃搅拌20min后,分批加入碳酸钠粉末795g至体系ph=8,加毕后保温20℃搅拌30min,再加入乙酸乙酯4800ml*1次萃取,水相继续用乙酸乙酯提取1620ml*2次,有机相合并后水洗3240ml*2次,分液,有机相用无水硫酸镁162g搅拌干燥2h,过滤,滤饼用乙酸乙酯淋洗162ml*3次,滤液合并后,负压浓缩(35℃,-0.09mpa,4h)至剩余糊状物约900g,加入石油醚1620ml,20℃打浆2h,抽滤,滤饼用石油醚淋洗162ml*2次,收集滤饼,真空干燥(25℃,-0.09mpa,约4h)至挥发份<2%,得到灰色固体粉末307g,纯度为97.8885%,收率为75.80%(以二氨基马来腈计);
步骤2,4-氨基-5-咪唑甲酰胺的合成:
氩气保护下,向装有温度计、回流冷凝管和尾气吸收装的3l三口瓶中加入水1450ml,氢氧化钠290g,290g中间体1,升温至95℃保温反应2h后,开始每隔1h取样分析,约3h反应完全,停止反应,自然降温至20℃。
后处理:体系进一步降温至0℃,控温0℃滴加盐酸(浓度35%),调体系至ph=6.5,滴毕搅拌1h,过滤,滤饼用100ml*2次水淋洗,滤饼抽干后烘料(50℃,常压,8h)至水分<15%,得到灰绿色固体粗品265g。
重结晶:氩气保护下,向2l的三口瓶中加入水1060ml,粗品265g,加热升温至60℃溶解,保温(60℃)搅拌30min,过砂芯漏斗,滤饼用50ml*2次的60℃热水淋洗,合并滤液,加入到2l三口瓶中,搅拌降温至3℃析晶1h,过滤,滤饼抽干后湿重约360g,加入无水乙醇400ml,-10℃~-15℃打浆2h,过滤,滤饼抽干,真空干燥(50℃,-0.09mpa)至水分<12.5%,共得到灰绿色固体粉末210g,4-氨基-5-咪唑甲酰胺纯度lc=99.8596%,收率为77.59%(以中间体1计),即为目标产物两步反应总收率为58.81%(以二氨基马来腈)。
实施例2
一种4-氨基-5-咪唑甲酰胺的工业化生产方法,包括以下步骤:
步骤1,中间体1的合成:
氩气保护条件下,向装有温度计、滴加漏斗和机械搅拌的5l的三口瓶中依次加入thf1458ml、二氨基马来腈162g,甲酰胺101.3g(1.5eq.),低温浴槽降温至0℃,开始滴加三氯氧磷344.7g(1.5eq.),滴毕后保温35℃反应2h,开始每隔1h取样,当二氨基马来腈lc<0.3%时,停止反应。
后处理:控温30℃,向反应液中滴加甲醇216g,滴加完毕控温30℃反应2h后,将体系缓慢倒入水2.6l中淬灭,淬灭完保温30℃搅拌20min后,分批加入碳酸钠粉末487g至体系ph=8.5,加毕后保温30℃搅拌30min,再加入乙酸乙酯2400ml*1次萃取,水相继续用乙酸乙酯提取810ml*2次,有机相合并后水洗1620ml*2次,分液,有机相用无水硫酸镁80g搅拌干燥2h,过滤,滤饼用乙酸乙酯淋洗80ml*3次,滤液合并后,负压浓缩(40℃,-0.08mpa,4h)至剩余糊状物约450g,加入石油醚810ml,30℃打浆2h,抽滤,滤饼用石油醚淋洗80ml*2次,收集滤饼,真空干燥(30℃,-0.08mpa,4h)至挥发份<2%,得到灰色固体粉末148g,纯度为97.8133%,收率为73.08%(以二氨基马来腈计);
步骤2同实施例1。
实施例1和实施例2近似,仅以实施例1表征结果为例进行相关说明,图1是实施例1制得的4-氨基-5-咪唑甲酰胺的lc图谱,由图1可得:得到的目标产物lc纯度99.8596%,产品lc含量高。图2是实施例1制得的4-氨基-5-咪唑甲酰胺的核磁氢谱图,由图2可得:δ11.1ppm~11.8ppm(d,1h,nh),7.0ppm~7.5ppm(d,1h,ch),6.5ppm~6.9ppm(m,2h,conh2),5.2ppm~5.8ppm(m,2h,c-nh2),符合目标产物的核磁氢谱图。
由此可得,本发明以二氨基马来腈为原料经两步反应成功合成了4-氨基-5-咪唑甲酰胺,所用的原材料简单,低毒,不会产生含氰根(剧毒)或含氮的废水产生,仅只会产生少量的含磷的废水,本发明的合成技术对环境相对友好;本发明常规反应设备即可满足工业化生产的要求,工厂也不需要特殊的资质;得到的产物纯度高达99.8%以上,且品质稳定,重复性好;由于合成路线所用的原料简单,价格低廉,且合成总收率能到58.81%(而背景技术中的收率最高为32%),故单位产品的原材料成本较低;由于合成路线仅为两步反应,且每步反应的反应时间及后处理时间均较短,后处理简单,故单位产品的制造成本较低;由于合成路线所用物料相对环保,不会产生大量的废水、废固及废气,故单位产品的环保成本低,综合考虑该产品的单位原材料成本、单位制造成本、单位环保成本,故该产品的综合成本较背景技术中剧毒的氰化物方法(氰化钠、氢氰酸、氰基丙醇)或者氨甲醇的合成方法均有着极为明显的优势,适合工业化生产。
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内也意图包含这些改动和变型在内。
- 还没有人留言评论。精彩留言会获得点赞!