一种T7核酸内切酶I的制备方法与流程
本申请是中国国家申请号201410141111.4,发明名称为“一种t7核酸内切酶i的制备方法”的分案申请。
本发明属于生物技术领域,具体涉及一种核酸内切酶的制备方法。
背景技术:
t7核酸内切酶i是一种多功能核酸酶,可特异识别并拆分重组中间体(holidayjunction,hj)交叉结构,随机切刻双链dna,特异识别并切割双链dna中的切刻和单碱基错配位点。
该酶蛋白是t7噬菌体基因3编码产物,由包含149个氨基酸残基的多肽链构成,两条肽链通过结构域交换形成同源二聚体,其催化中心由来自两个亚基的氨基酸侧链共同组成,两个催化结构域可以独立作为dna切刻酶;通过结构域之间的协同作用,表现出结构特异性催化。作为一种已知的噬菌体类解离酶,该酶可以剪切分叉dna,随机切刻线性dna,识别并切刻nicked-dna位点,识别单碱基错配位点,t7核酸内切酶i对hj的结合具有高度特异性,其结合hj的能力是结合线性双链dna的1000倍左右,还具备随机在双链dna中引入切刻位点的活性,专一性识别切刻位点并在另一条dna链上对应位置引入另一切刻位点导致dna双链的断裂,专一性识别并切割dna双链中的单碱基错配位点。该酶应用广泛,具有作为工具酶的重要价值。主要的商业用途包括:可用于分支dna结构的解离,基因组测序技术和dna-shuffling技术中dna的前处理,snp的检测等。
目前制备t7核酸内切酶的方法是:将蛋白分别表达合成出两段肽链,在体外融合而成。采用impact-twin蛋白纯化系统纯化与mbp融合表达的截短型无活性t7核酸内切酶i(tt7endoi),该融合蛋白的c-端带有硫酯键。然后体外合成一段合成肽,其中含有被前者截下来的氨基酸,将该合成肽与融合蛋白的c-端硫酯键相连接,形成全长具有活性的t7核酸内切酶i。最后,再用因子xa将mbp融合头切去,纯化得到全长的野生型t7核酸内切酶i。
技术实现要素:
本发明的目的在于提供一种核酸内切酶的制备方法,该方法操作简便、易于规模化生产,并能有效节约成本。
本发明所述的一种t7核酸内切酶的制备方法,包括以下步骤:
(a)t7核酸内切酶的表达载体的构建;
(b)将不含有终止密码子的编码序列插入到步骤a所获得的蛋白表达载体中;以及
(c)采用与标签相适应的纯化方法,获得目标蛋白质即t7核酸内切酶。
根据本发明所述的t7核酸内切酶的制备方法的进一步特征,所述表达载体包括:用于插入目的蛋白即t7核酸内切酶的编码序列的识别和切割位点;在所述识别和切割位点的上游具有如seqidno:1所示的giiis信号肽编码序列,其前面是起始密码子;在所述识别和切割位点的下游具有his6标签,其后面是终止密码子。
优选地,所述的识别和切割位点为bsai识别及切割位点。
本发明的优点在于:构建n端带m13噬菌体蛋白iii的前导序列(giiis)的载体,合成出来的信号肽能引导蛋白分泌出核外,在细胞周质中表达,对细胞本身的基因组无毒性,故能直接在体内表达出可溶并有生物活性的t7核酸内切酶i。这种生产方法简便易操作,易于规模化生产,并能有效节约成本。
经实验表明,本发明所述方法获得的t7核酸内切酶i能特异地识别dna片段中不匹配的位点,并在这此位点附近进行切割。其切割效率与商品化的t7核酸内切酶i相当,活性良好。相比于现有技术,本发明所述方法操作更为简便、易于规模化生产,并能有效节约成本。
附图说明
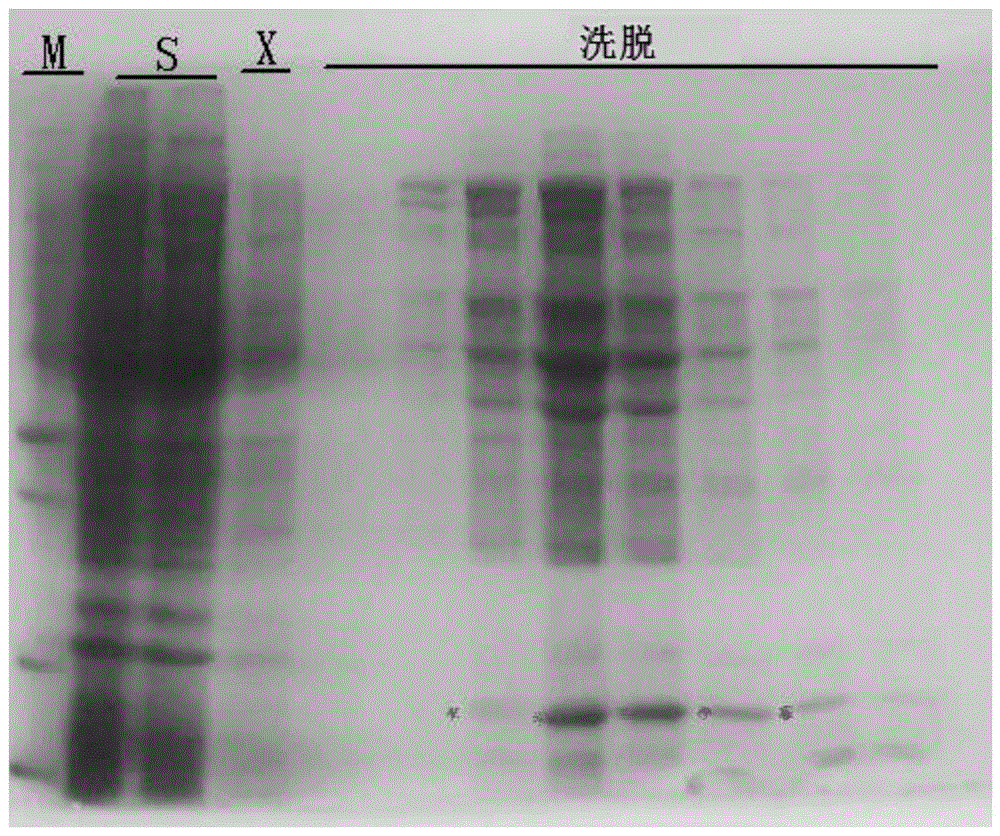
图1是t7核酸内切酶i的ni柱层析电泳图;图中,s:层析前样品;x:穿过样品;elution:线性洗脱收集峰。
图2是t7核酸内切酶i的heparin柱层析电泳图;图中,s:层析前样品;x:穿过样品;elution:线性洗脱收集峰。
图3是t7核酸内切酶i的hitrapspp柱层析电泳图;图中,s:层析前样品;x:穿过样品;elution:线性洗脱收集峰。
图4是反应产物用于琼脂糖凝胶电泳图;图中,m:100-3000bpdnamarker;1:加入nebt7核酸内切酶i(nebcatalog#m0302)的阳性对照;2:加入纯化后的t7核酸内切酶i;3:不加任何酶,加入ddh2o作为阴性对照;圆点:t7核酸内切酶i识别后切开所形成的条带,长度分别在480bp及330bp左右。
具体实施方式
以下合通过具体实施例结合附图的方式对本发明做进一步说明,目的在于本发明的内容而非限制发明的保护范围。下面实施例中,除非特别注明试验方法的具体条件,通常按照常规条件,如sambrook等人著的《分子克隆:试验指南》(newyork,coldspringhabourlaboratorypress)中所述条件,或按照制造厂商建议条件进行。实验所需菌种、质粒、试剂等均为商品化产品,均可通过生物制品厂商购买获得。
实施例1:t7核酸内切酶i的制备
制备方法:将t7基因的编码序列构建表达克隆,于大肠杆菌中表达,重组蛋白经过层析纯化后,获得目标蛋白。
具体流程如下:
(1)构建带bsai酶切位点的pet28-giiis表达载体
在pet28表达载体xhoi酶切位点上游加入一个bsai识别及切割位点(5’-ggtctct/aggt-3’),并在bsai位点的上游加上起始密码子atg和giiis信号肽编码序列,信号肽氨基酸序列如seqidno:1所示。
seqidno:1kkllfaiplvvpfyshs
(2)构建含基因片段的表达克隆
设计含有bsai酶切位点的上游引物(seqidno:2)和xhoi(seqidno.3)酶切位点的下游引物,从大肠杆菌t7噬菌体基因组中进行pcr扩增,获得459bp含有t7核酸内切酶i基因编码序列的插入片段,其开放阅读框中不含有终止密码子。用bsai和xhoi两个内切酶对片段及载体进行酶切,酶切之后的片段与载体含有相同的粘性末端。
seqidno:25’-atatatggtctcgtagcatggcaggttacggcgct-3’
seqidno.35’-atatatctcgagtttctttcctcctttcctttttaa-3’
用t4dna连接酶连接插入片段和pet28载体。插入片段开放阅读框的下游包含有一个his6标签,以用于下游ni柱纯化。his6标签后含有一个终止密码子tga。连接产物进行转化到e.coli2t1感受态,在lb培养基上37℃过夜培养。通过鉴定后筛选出正确的转化子在lb液体培养基中扩大培养。
(3)收集菌液,细胞沉淀悬浮缓冲液a(20mmtris、50mmnacl、2.5%甘油,ph7.0)和2xotg等体积混合的溶液中,然后以大约18000psig通过高压破碎机,裂解物于12000rmp离心40min后收集上清。上清按1000∶1的比例加入polyethylenimine充分混匀30分钟,12000rmp离心40分钟收集上清过0.22μm滤膜。
(4)将上清加到已用缓冲液a平衡好的histrap-ni柱上。用缓冲液a冲平基线,然后用0-500mm的imidazole线性梯度洗脱收集。对各收集组份进行sds-page检测(图1),收集含目的蛋白的组份。
(5)含目的蛋白的组份被汇集到一起,将样品上样到已经平衡好的heparin柱子上。用缓冲液a冲平基线,然后用0-1m的nacl线性洗脱。对各收集组份进行sds-page检测(图2),收集含目的蛋白的组份。
(6)将上述各组份汇集到一起透析到缓冲液a后,样品上样到已用缓冲液a平衡好的hitrapspp柱子上。用缓冲液a冲平基线,然后用0-1m的nacl线性洗脱。对各收集组份进行sds-page检测(图3),收集含目的蛋白的组份。
(7)将上述各组份汇集到一起透析到贮存缓冲液(200mmnacl、20mmtris-hclph7.5、0.01mmedta、1mmdtt、0.15%tritonx-100、50%glycerin)。纯化后的酶在-20℃保存。
实施例2:t7核酸内切酶i的活性检测
设计两个含有10个碱基差异的克隆,取相同的上游和下游引物进行扩增,得到一对有10个碱基不匹配的pcr产物。序列见seqidno:4及seqidno:5。
将两种pcr产物等量混合,在含有1x缓冲液(50mmnacl,10mmtris-hcl,10mmmgcl21mmdtt,ph7.9@25℃)中95℃变性5min,再退火至室温,形成具有一个重组中间体(hj)的产物。在上述退火体系中加入1u实施例1所制备的t7核酸内切酶i,37℃孵育1小时。以nebt7核酸内切酶i(catalog#m0302)为阳性对照(pc),并设置不加酶的阴性对照(nc)。反应结束后加入含有sds的上样缓冲液终止反应。反应产物用于琼脂糖凝胶电泳,结果见图4。
结果分析表明:本发明所述方法得到的t7核酸内切酶i能特异地识别dna片段中不匹配的位点,并在这此位点附近进行切割。其切割效率与商品化的t7核酸内切酶i相当,证明其活性保持良好。
- 还没有人留言评论。精彩留言会获得点赞!