一种工业大麻性状相关基因快速定位方法与流程
本发明属于工业大麻研究技术领域,具体涉及一种工业大麻性状相关基因快速定位方法。
背景技术:
工业大麻(cannabissativa.l)为大麻科(cannabinaceae)大麻属(cannabis)一年生草本植物,其四氢大麻酚(thc)含量低于0.3%,工业大麻的茎秆、花叶和籽粒等都有经济利用价值,在纺织、建筑、造纸、医药和食品等产业有广泛应用,工业大麻品种培育的主要目标可根据特定产业需求来制定,目前,工业大麻育种主要采用传统育种方法,但该方法培育新品种不仅周期长而且很难达到定制的育种目标,通过分子育种方法不仅能提高育种效率而且可以达到精准育种的目的,但前提是对性状相关基因进行定位寻找可靠分子标记,但由于工业大麻分子生物学研究起步较晚,相关功能基因尚未定位,测序技术及生物信息学的发展为性状基因的快速定位提供可能。
技术实现要素:
本发明提供一种工业大麻性状相关基因快速定位方法,解决了目前工业大麻分子生物学起步晚相关功能基因尚未定位等问题。
为实现上述目的,本发明采取的技术方案如下:
一种工业大麻性状相关基因快速定位方法,所述方法步骤包括:根据目标性状进行品种选择,极端群体构建,特异位点扩增片段(slaf)文库构建及高通量测序,标记开发,关联分析,基因注释和实时荧光定量pcr。
进一步地,所述品种选择具体为:选择目标性状差异显著,其他性状(包括农艺、产量和抗病性等)田间表现较为一致的两个品种作为父母本。
进一步地,所述极端群体构建具体为:根据花期选择合适时间种植两个品种,在现蕾期拔除母本品种雄株或去除雄花,获得的杂交种即为f1代,在现蕾期选择性别相同的工业大麻植株进行挂牌,取幼嫩叶片液氮速冻,-80℃冰箱保存待用,在工艺成熟期或者籽粒成熟期收获工业大麻,测量目标性状数据。
进一步地,所述特异位点扩增片段文库构建及高通量测序具体为:根据目标性状统计结果将样本分成差异显著的两组,每组至少选择30-100株植株(约占样本总量的5%,如1000株植株则每组选择50株植株),提取dna后,每个植株样本dna等量混合构建高低两个混池,用酶切测序软件slaf-predict对工业大麻参考基因组进行电子酶切预测,使用内切酶对dna混池进行酶切,对酶切片段进行回收,在3'端加a,连接dual-index测序接头,对各个样本中同一位点处的dna序列片段进行pcr扩增、纯化、混和样品和切胶,经slaf文库检验合格后,扩增产物用测序系统进行测序,对测序结果用bwa软件以水稻(oryzasativa)作为对照,评估酶切方案是否有效。
进一步地,所述标记开发具体为:对获得的reads进行聚类分析,对工业大麻各染色体上的slaf标签的分布进行作图,用gatk软件包对cleanreads在参考基因组上的定位结果进行snp检测。
进一步地,所述关联分析具体为:在关联分析之前,首先对snp位点进行过滤,过滤掉read支持度小于4的位点、具有多个基因型的位点、隐性混池基因不是来自于隐性亲本的位点及混池之间基因型一致的位点;采用snp-index方法进行关联分析寻找混池之间基因型频率的显著差异,用δ(snp-index)进行统计,snp与目标性状关联度越强,δ(snp-index)的数值越接近于1;计算公式如下:snpindex(aa)=maa/(maa+paa);snpindex(ab)=mab/(mab+pab);δ(snp-index)=snpindex(aa)–snpindex(ab);
注:paa是指aa池来源于父本的深度,maa是指aa池来源于母本的深度,pab是指ab池来源于父本的深度,mab是指ab池来源于母本的深度;
假阳性位点的消除主要利用标记在基因组上的位置,采用snpnum方法对δsnp-index进行拟合后,根据关联阈值,在阈值以上的区域选择作为性状相关候选区域,并根据计算机模拟实验计算结果。
进一步地,所述基因注释具体为:对关联分析得到的候选区域内的编码基因进行nr、swiss-prot、go、kegg和cog的深度注释,通过注释结果快速筛选候选基因。
进一步地,所述实时荧光定量pcr具体为:选择目标性状有差异的2-3个品种,提取样本rna,逆转录,针对不同候选基因各自设计1-2对相应的引物进行引物调试,调试结果合格的用于相对定量pcr分析。
本发明相对于现有技术的有益效果为:对目标性状差异显著的亲本通过杂交获得的f1代群体构建高低混池,对亲本进行重测序,对高低混池采用简化基因组深度测序技术(specificlocationamplifiedfragmentssequence,slaf-seq)进行测序,开发slaf标签及进行单核苷酸多态性(snp)检测,通过对混池间的基因型频率差异进行标记关联分析,获得目标性状相关候选区域,多数植物群体至少要扩繁至f2代才能进行基因定位,由于工业大麻的高杂合度特性,通过f1代即可实现基因定位,该方法证明能以高效低成本的方式实现工业大麻基因的快速定位。
附图说明
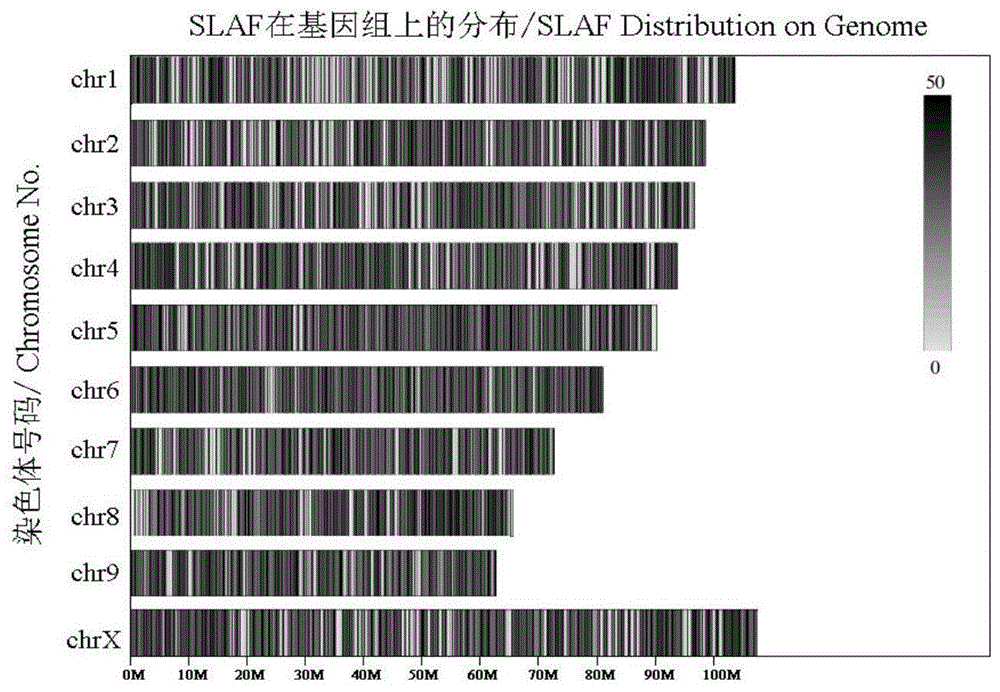
图1为slaf标签(黑色线)在工业大麻染色体上的分布图;
图2为snp-index关联值在染色体上的分布图;
注:横坐标为染色体名称,黑色的点代表计算出来的snp-index(或δsnp-index)值,黑色的线为拟合后的snp-index(或δsnp-index)值。上图是隐性混池的snp-index值的分布图;中图是显性混池的snp-index值的分布图;下图是δsnp-index值的分布图,其中虚线代表99百分位数的阈值线。
图3为候选区域内基因的通路分布图;
图4为基因loc115705530荧光定量pcr结果图(*p<0.05,**p<0.01);
图5为基因loc115707511荧光定量pcr结果图(*p<0.05,**p<0.01);
图6为基因loc115704794荧光定量pcr结果图(*p<0.05,**p<0.01);
图7为基因loc115705371荧光定量pcr结果图(*p<0.05,**p<0.01);
图8为基因loc115705688荧光定量pcr结果图(*p<0.05,**p<0.01)。
具体实施方式
下面结合附图和实施例对本发明的技术方案作进一步的说明,但并不局限于此,凡是对本发明技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,均应涵盖在本发明的保护范围中。
实施例1:
(1)品种选择:选择高纤维含量的金刀-15为父本,低纤维含量的火麻一号为母本进行杂交。
(2)群体构建:金刀-15和火麻一号花期一致,在同一时间种植两个品种,在现蕾期拔除火麻一号雄株,获得的杂交种即为f1代,在现蕾期选择性别相同的工业大麻植株进行挂牌,取幼嫩叶片液氮速冻,-80℃冰箱保存待用,在工艺成熟期收获工业大麻,麻株经鲜剥皮后测量单株原茎重量和纤维重量,计算纤维含量。计算公式如下:纤维含量=纤维重量/原茎重量×100%。
(3)slaf文库构建及高通量测序:根据纤维含量结果选择30株含量高的植株和30株含量低的植株,提取dna后,每个植株样本dna等量混合构建高低两个混池,用酶切测序软件slaf-predict对工业大麻参考基因组进行电子酶切预测可得105823个slaf标签,各标签在基因组染色体上分布基本均匀,如图1所示,选择rsai和haeiii对dna混池进行酶切,对酶切片段进行回收,在3'端加a,连接dual-index测序接头,对各个样本中同一位点处的dna序列片段进行聚合酶链式反应(polymerasechainreaction,pcr)扩增、纯化、混和样品和切胶,经slaf文库检验合格后,扩增产物用测序系统illuminahiseqtm2500进行测序。对测序结果用bwa软件以水稻(oryzasativa)作为对照,评估酶切方案是否有效。
(4)标记开发:对获得的reads进行聚类分析,对工业大麻各染色体上的slaf标签的分布进行作图,用gatk软件对cleanreads在参考基因组上的定位结果进行snp检测,混池间检测到snp位点389,687个,可进行snp分子标记开发。
(5)关联分析:在关联分析之前,首先对snp位点进行过滤,过滤掉read支持度小于4的位点、具有多个基因型的位点、隐性混池基因不是来自于隐性亲本的位点及混池之间基因型一致的位点。采用snp-index方法进行关联分析,主要目的是寻找混池之间基因型频率的显著差异,用δ(snp-index)进行统计。snp与目标性状关联度越强,δ(snp-index)的数值越接近于1。计算公式如下:snpindex(aa)=maa/(maa+paa);snpindex(ab)=mab/(mab+pab);δ(snp-index)=snpindex(aa)–snpindex(ab)
注:paa是指aa池来源于父本的深度,maa是指aa池来源于母本的深度,pab是指ab池来源于父本的深度,mab是指ab池来源于母本的深度;
假阳性的位点的消除主要利用标记在基因组上的位置,采用snpnum方法对δsnp-index进行拟合后,根据关联阈值,在阈值以上的区域选择作为性状相关候选区域,并根据计算机模拟实验计算结果。当置信度为0.90时没有关联到相关候选区域。理论上,目标位点及其附近的连锁位点应该接近于该阈值,显著关联区域附近应该出现一个较高的峰值,但在本实验结果中因为没有发现超过理论阈值的区域,因此没有显著的定位结果。为了充分发掘利用数据,通过降低阈值来寻找可能的定位区域,利用拟合后δsnp-index的99百分位数,即0.10,如图2所示。共得到总长度为8.72mb的4个候选区域,共包括397个基因。
(6)基因注释:对关联分析得到的候选区域内的编码基因进行nr、swiss-prot、go、kegg和cog的深度注释,共注释到389个基因,候选区域内基因的通路分布图如图3所示,通过与拟南芥、亚麻和棉花等作物进行基因比对,获得候选基因loc115705530、loc115707511、loc115703881、loc115704794、loc115705010、loc115705371、loc115705568、loc115705688、loc115705891、loc115705892和loc115706200。
(7)实时荧光定量pcr:选择纤维含量不同的3个品种火麻一号(22.1%)、汉麻10号(27.1%)和金刀15(33.1%)的苗期和工艺成熟期进行验证,提取样本rna(具体方法参照天根植物rna提取试剂盒(dp432)),使用fastkingrtkit(kr116)试剂进行反转录,体系20ul,配置buffer混合液(2ulfq-rtprimermix,2ul10×kingrtbuffer,1ulfastkingrtenzymemix,5ulrnase-freeddh2o),1ugrna加buffer混合液10ul,用ddh2o补充至20ul,42℃30min,95℃3min程序下进行反应,反转录出的cdna10倍稀释后用于定量pcr,针对不同候选基因各自设计1-2对相应的引物进行引物调试,调试结果合格的可用于定量pcr分析,用试剂powerqpcrpremix(genecopoeia)进行定量分析,96孔板sybrgreen20ul体系(10ulmix,1ulcdna,前后引物各0.5ul,8ulh2o,反应程序如下表所示。
采用标准曲线法,构建目的基因和内参基因的标准品,构建标准曲线,计算目的基因和内参基因引物的扩增效率,代入计算得出两者的倍数关系,拷贝数计算公式:拷贝数/μl=(ng/μl)×10-9×6.02×1023/(bp×660)。其中:6.02×1023为摩尔常数,660为碱基(agct)平均分子量。通过定量结果发现基因loc115705530、loc115707511、loc115704794、loc115705371和loc115705688与纤维含量相关,如图4-8所示。
- 还没有人留言评论。精彩留言会获得点赞!