一种环烯烃化合物及其制备方法和应用与流程
1.本发明属于有机合成技术领域,具体涉及一种环烯烃化合物及其制备方法和应用,尤其涉及一种耐热性好的环烯烃化合物及其制备方法和应用。
背景技术:
2.双环戊二烯和降冰片烯是最为常用的环烯烃单体,具有较大的环张力,在开环易位聚合或者是加成共聚反应中表现出较高的活性。最常见的环烯烃共聚物是乙烯/降冰片烯共聚物。
3.此类环烯烃材料有很多优异的性能,但是也存在一定的不足,环烯烃共聚物的玻璃化转变温度随着环烯烃单体的插入率的增大而呈现上升趋势,但是高的环烯烃单体插入率常常会带来材料韧性下降的问题,这一缺点导致了乙烯/降冰片烯共聚物的应用受到一定限制。从相关报道可知,当降冰片烯的插入率高于54mol%时,乙烯/降冰片烯共聚物的玻璃化转变温度才能够达到150℃,与 pc的玻璃化转变温度类似,但此时共聚物材料的脆性大,韧性下降明显。环烯烃单体的结构及其插入率决定着共聚物的性能,因此开发新的环烯烃单体对改善或者提高环烯烃共聚物的性能具有至关重要的作用。
4.cn101319020报道了一种应用于光学材料、偏振片以及液晶显示器的环烯烃聚合物,通过采用两种单体共聚,制得含及的环烯烃聚合物,但其所用的单体结构较复杂,原料不易得,同时没有证据表明其耐热性有所提高。
5.因此突破特殊结构的环烯烃单体的合成工艺,将为环烯烃共聚物材料提供关键原材料及重要的保障。
技术实现要素:
6.针对现有技术的不足,本发明的目的在于提供一种环烯烃化合物及其制备方法和应用,尤其提供一种耐热性好的环烯烃化合物及其制备方法和应用。
7.为达到此发明目的,本发明采用以下技术方案:
8.第一方面,本发明提供一种环烯烃化合物,所述环烯烃化合物的结构如式 (ⅰ)所示:
9.10.其中,r1、r2、r3独立地选自h或ch3;k选自0或1。
11.本发明所涉及的上述环烯烃单体是一种含单桥或者双桥环酯基的降冰片烯衍生物,其取代基为具有单桥或者双桥环的酯基,其特殊的结构赋予单体较强的刚性及空间位阻;与常见的环烯烃化合物如降冰片烯、双环戊二烯相比,具有更大体积的取代基,与乙烯共聚后,在相同环烯烃单体的嵌入率下,共聚物将表现出更高的玻璃化转变温度,更好的耐热性。
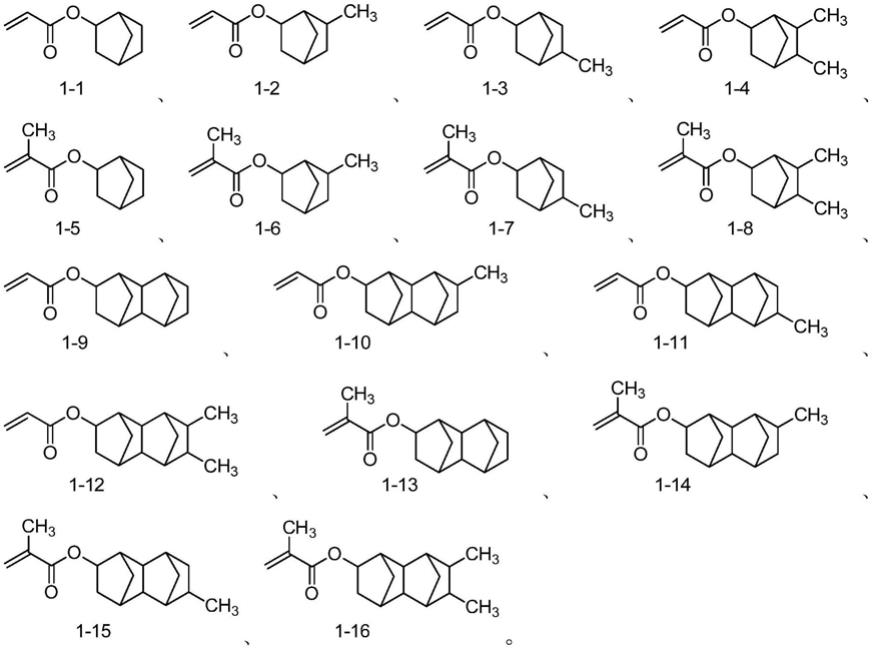
12.在本发明中,所述环烯烃化合物的结构选自如下式(1)
‑
(16)的结构:
[0013][0014]
第二方面,本发明提供如第一方面所述的环烯烃化合物的制备方法,所述制备方法包括:将环戊二烯和/或双环戊二烯、式(ⅱ)所示化合物、阻聚剂混合,进行diels
‑
alder加成反应,制得所述环烯烃化合物;
[0015][0016]
其中,r1、r2、r3独立地选自h或ch3;k选自0或1。
[0017]
上述合成工艺转化率高,制备得到的环烯烃化合物纯度高。该合成工艺的示意如下所示,当反应原料为双环戊二烯时,其在高温下经过裂解生成环戊二烯,进一步地与(甲
基)丙烯酸酯反应可生成目标物,所述反应原料双环戊二烯可不用单独进行裂解,可以直接与(甲基)丙烯酸酯混合进行反应,同样可获得目标物。所述(甲基)丙烯酸酯本领域技术人员可以通过商购或者依照现有技术合成得到,所述现有技术示例性地可以是cn108299194a中公开的方法。
[0018][0019]
在本发明中,所述式(ⅱ)所示化合物的结构选自如下式(1
‑
1)
‑
(1
‑
16) 的结构:
[0020]020][0021]
优选地,所述环戊二烯和/或双环戊二烯中环戊二烯单体与式(ⅱ)所示化合物的摩尔比为(1
‑
2):1,例如1:1、1.2:1、1.5:1、1.8:1或2:1等,上述范围内的其他具体点值均可选择,在此便不再一一赘述。
[0022]
优选地,所述diels
‑
alder加成反应的温度为170
‑
230℃,例如170℃、180℃、190℃、200℃、210℃、220℃或230℃等,上述范围内的其他具体点值均可选择,在此便不再一一赘述。所述diels
‑
alder加成反应的时间为1
‑
5h,例如1h、 2h、3h、4h或5h等,上述范围内的其他具体点值均可选择,在此便不再一一赘述。
[0023]
所述环戊二烯为双环戊二烯在170℃或更高温度下的裂解产物,因反应温度为170
‑
230℃,在该温度条件下,双环戊二烯自身会发生裂解生成环戊二烯,因此优选双环戊二烯为反应原料。
[0024]
优选地,所述diels
‑
alder加成反应的压力为0.3
‑
2.0mpa,例如0.3mpa、 0.5mpa、0.8mpa、1.0mpa、1.2mpa、1.5mpa、1.8mpa或2.0mpa等,上述范围内的其他具体点值均可选择,在此便不再一一赘述。
[0025]
优选地,所述反应结束后进行减压蒸馏。
[0026]
优选地,所述减压蒸馏的温度150
‑
200℃,例如150℃、160℃、170℃、180℃、 190℃或200℃等,真空度为10
‑
500pa,例如10pa、50pa、100pa、200pa、 230pa、300pa、350pa、400pa或500pa等,上述范围内的其他具体点值均可选择,在此便不再一一赘述。
[0027]
优选地,所述阻聚剂包括吩噻嗪、对羟基苯甲醚或对苯二酚。
[0028]
优选地,所述阻聚剂的添加量为反应原料总投料质量的0.001%
‑
0.1%,例如0.001%、0.002%、0.005%、0.01%、0.02%、0.05%、0.06%、0.08%或0.1%等,上述范围内的其他具体点值均可选择,在此便不再一一赘述。
[0029]
第三方面,本发明提供一种如上所述的环烯烃化合物在制备环烯烃共聚物材料中的应用,所述环烯烃共聚物材料由环烯烃化合物与α
‑
烯烃加成聚合制得。
[0030]
所述环烯烃单体与乙烯经加成聚合可制备环烯烃共聚物,共聚物具有优异的耐热性。
[0031]
与现有技术相比,本发明具有如下有益效果:
[0032]
本发明所涉及的环烯烃化合物是一种含单桥或者双桥环酯基的降冰片烯衍生物,其取代基为具有单桥或者双桥环的酯基,其特殊的结构赋予单体较强的刚性及空间位阻;与常见的环烯烃化合物如降冰片烯、双环戊二烯相比,具有更大体积的取代基,与乙烯共聚后,在相同环烯烃单体的嵌入率下,共聚物将表现出更高的玻璃化转变温度,更好的耐热性。
[0033]
本发明所涉及的环烯烃化合物的制备方法转化率高,制备得到的环烯烃化合物纯度高。
具体实施方式
[0034]
为更进一步阐述本发明所采取的技术手段及其效果,以下结合本发明的优选实施例来进一步说明本发明的技术方案,但本发明并非局限在实施例范围内。
[0035]
实施例1
[0036]
本实施例提供制备一种环烯烃化合物其制备方法如下:
[0037]
将双环戊二烯(66g,0.5mol)、(166g,1.0mol)及吩噻嗪(0.0232 g)投于反应釜,抽真空通氮气,反复操作3次置换釜内空气,保持釜内压力0.1 mpa,开启加热
及搅拌,升温至170℃,反应1h后,冷却出料,对反应结束液进行减压蒸馏,收集蒸馏温度150℃,真空度200pa下的馏分,得到无色透明液体,即为目标产物(1)
[0038]
对制得的产品进行如下表征:
[0039]
气相色谱分析gc:gc
‑
2014c,日本岛津公司,柱子db
‑
5,经测试,产品含量为98.50%。
[0040]
元素分析:ce
‑
440型有机碳氢氮氧硫元素分析仪,美国加联公司,经测试,元素为c:77.56%(77.55%),h:8.66%(8.68%)。
[0041]1h
‑
nmr(cdcl3为氘代试剂,四甲基硅烷为内标):6.23ppm(2h),3.90ppm(1h),3.77ppm(1h),2.54ppm(1h),2.37ppm(1h),2.27ppm(1h), 2.13ppm(1h),1.99ppm(1h),1.95ppm(1h),1.88ppm(1h),1.74ppm(2h), 1.70ppm(1h),1.50ppm(1h),1.58ppm(2h),1.44ppm(1h),1.33ppm(2h)。
[0042]
实施例2
[0043]
本实施例提供制备一种环烯烃化合物其制备方法如下:
[0044]
将双环戊二烯(79.2g,0.6mol)、(180g,1.0mol)及吩噻嗪(0.1056g)投于反应釜,抽真空通氮气,反复操作3次置换釜内空气,通入氮气至釜内压力0.5mpa,开启加热及搅拌,升温至200℃,反应3h后,冷却出料,对反应结束液进行减压蒸馏,收集蒸馏温度180℃,真空度150pa下的馏分,得到无色透明液体,即为目标产物(2)
[0045]
对制得的产品进行如下表征:
[0046]
气相色谱分析gc:gc
‑
2014c,日本岛津公司,柱子db
‑
5,经测试,产品含量为98.2%。
[0047]
元素分析:ce
‑
440型有机碳氢氮氧硫元素分析仪,美国加联公司,经测试,元素为c:78.02%(78.01%),h:9.01%(9.00%)。
[0048]1h
‑
nmr(cdcl3为氘代试剂,四甲基硅烷为内标):6.23ppm(2h), 3.77ppm(1h),3.90ppm(1h),2.37ppm(1h),2.27ppm(1h),2.13ppm(1h), 2.02ppm(2h),1.95ppm(1h),1.88ppm(1h),1.75ppm(2h),1.70ppm(1h), 1.60ppm(1h),1.50ppm(2h),1.44ppm(1h),1.24ppm(1h),0.96ppm(3h)。
[0049]
实施例3
[0050]
本实施例提供制备一种环烯烃化合物其制备方法如下:
[0051]
将双环戊二烯(132g,1.0mol)、(180g,1.0mol)及吩噻嗪(0.0624g)投于反应釜,抽真空通氮气,反复操作3次置换釜内空气,通入氮气至釜内压力1.5mpa,开启加热及搅拌,升温至230℃,反应5h后,冷却出料,对反应结束液进行减压蒸馏,收集蒸馏温度180℃,真空度150pa下的馏分,得到无色透明液体,即为目标产物(3)
[0052]
对制得的产品进行如下表征:
[0053]
气相色谱分析gc:gc
‑
2014c,日本岛津公司,柱子db
‑
5,经测试,产品含量为98.5%。
[0054]
元素分析:ce
‑
440型有机碳氢氮氧硫元素分析仪,美国加联公司,经测试,元素为c:78.02%(78.01%),h:9.01%(9.00%)。
[0055]1h
‑
nmr(cdcl3为氘代试剂,四甲基硅烷为内标):6.23ppm(2h), 3.77ppm(1h),3.90ppm(1h),2.37ppm(1h),2.27ppm(1h),2.13ppm(1h), 2.02ppm(2h),1.95ppm(1h),1.88ppm(1h),1.75ppm(2h),1.70ppm(1h), 1.60ppm(1h),1.50ppm(2h),1.44ppm(1h),1.24ppm(1h),0.96ppm(3h)。
[0056]
实施例4
[0057]
本实施例提供制备一种环烯烃化合物其制备方法如下:
[0058]
将双环戊二烯(79.2g,0.6mol)、(194g,1.0mol)及吩噻嗪 (0.0520g)投于反应釜,抽真空通氮气,反复操作3次置换釜内空气,通入氮气至釜内压力2.0mpa,开启加热及搅拌,升温至170℃,反应1h后,冷却出料,对反应结束液进行减压蒸馏,收集蒸馏温度190℃,真空度200pa下的馏分,得到无色透明液体,即为目标产物(4)
[0059]
对制得的产品进行如下表征:
[0060]
气相色谱分析gc:gc
‑
2014c,日本岛津公司,柱子db
‑
5,经测试,产品含量为98.3%。
[0061]
元素分析:ce
‑
440型有机碳氢氮氧硫元素分析仪,美国加联公司,经测试,元素为c:78.40%(78.42%),h:9.30%(9.29%)。
[0062]1h
‑
nmr(cdcl3为氘代试剂,四甲基硅烷为内标):6.23ppm(2h), 3.90ppm(1h),3.77ppm(1h),2.37ppm(1h),2.13ppm(1h),2.27ppm(1h), 2.02ppm(3h),1.88ppm(1h),1.70ppm(3h),1.50ppm(1h),1.59ppm(2h), 1.43ppm(1h),0.90ppm(6h)。
[0063]
实施例5
[0064]
本实施例提供制备一种环烯烃化合物其制备方法如下:
[0065]
将双环戊二烯(99.0g,0.75mol)、(180g,1.0mol)及吩噻嗪 (0.2790g)投于反应釜,抽真空通氮气,反复操作3次置换釜内空气,通入氮气至釜内压力2.0mpa,开启加热及搅拌,升温至230℃,反应5h后,冷却出料,对反应结束液进行减压蒸馏,收集蒸馏温度150℃,真空度50pa下的馏分,得到无色透明液体,即为目标产物(5)
[0066]
对制得的产品进行如下表征:
[0067]
气相色谱分析gc:gc
‑
2014c,日本岛津公司,柱子db
‑
5,经测试,产品含量为98.7%。
[0068]
元素分析:ce
‑
440型有机碳氢氮氧硫元素分析仪,美国加联公司,经测试,元素为c:78.00%(78.01%),h:8.98%(9.00%)。
[0069]1h
‑
nmr(cdcl3为氘代试剂,四甲基硅烷为内标):6.23ppm(2h), 3.90ppm(1h),2.89ppm(1h),2.54ppm(1h),2.27ppm(1h),2.13ppm(1h), 1.95ppm(2h),1.88ppm(1h),1.70ppm(3h),1.50ppm(1h),1.58ppm(2h), 1.44ppm(1h),1.33ppm(5h)。
[0070]
实施例6
[0071]
本实施例提供制备一种环烯烃化合物其制备方法如下:
[0072]
将双环戊二烯(79.2g,0.6mol)、(194g,1.0mol)及吩噻嗪(0.0546g)投于反应釜,抽真空通氮气,反复操作3次置换釜内空气,通入氮气至釜内压力0.5mpa,开启加热及搅拌,升温至170℃,反应1h后,冷却出料,对反应结束液进行减压蒸馏,收集蒸馏温度180℃,真空度500pa下的馏分,得到无色透明液体,即为目标产物(6)
[0073]
对制得的产品进行如下表征:
[0074]
气相色谱分析gc:gc
‑
2014c,日本岛津公司,柱子db
‑
5,经测试,产品含量为98.5%。
[0075]
元素分析:ce
‑
440型有机碳氢氮氧硫元素分析仪,美国加联公司,经测试,元素为c:78.44%(78.42%),h:9.28%(9.29%)。
[0076]1h
‑
nmr(cdcl3为氘代试剂,四甲基硅烷为内标):6.23ppm(2h), 3.90ppm(1h),2.89ppm(1h),2.27ppm(1h),2.13ppm(1h),2.02ppm(3h), 1.88ppm(1h),1.70ppm(3h),1.60ppm(1h),1.44ppm(3h),1.38ppm(3h), 1.23ppm(1h),0.96ppm(3h)。
[0077]
实施例7
[0078]
本实施例提供制备一种环烯烃化合物其制备方法如下:
[0079]
将双环戊二烯(79.2g,0.6mol)、(194g,1.0mol)及吩噻嗪(0.0546g)投于反应釜,抽真空通氮气,反复操作3次置换釜内空气,通入氮气至釜内压力0.5mpa,开启加热及搅拌,升温至230℃,反应3h后,冷却出料,对反应结束液进行减压蒸馏,收集蒸馏温度180℃,真空度450pa下的馏分,得到无色透明液体,即为目标产物(7)
[0080]
对制得的产品进行如下表征:
[0081]
气相色谱分析gc:gc
‑
2014c,日本岛津公司,柱子db
‑
5,经测试,产品含量为98.5%。
[0082]
元素分析:ce
‑
440型有机碳氢氮氧硫元素分析仪,美国加联公司,经测试,元素为c78.40%(78.42%),h:9.30%(9.29%)。
[0083]1h
‑
nmr(cdcl3为氘代试剂,四甲基硅烷为内标):6.23ppm(2h), 3.90ppm(1h),2.89ppm(1h),2.27ppm(1h),2.13ppm(1h),2.02ppm(3h), 1.88ppm(1h),1.70ppm(3h),1.60ppm(1h),1.44ppm(3h),1.38ppm(3h), 1.23ppm(1h),0.96ppm(3h)。
[0084]
实施例8
[0085]
本实施例提供制备一种环烯烃化合物其制备方法如下:
[0086]
将双环戊二烯(79.2g,0.6mol)、(208g,1.0mol)及吩噻嗪(0.0574g)投于反应釜,抽真空通氮气,反复操作3次置换釜内空气,通入氮气至釜内压力1.0mpa,开启加热及搅拌,升温至200℃,反应5h后,冷却出料,对反应结束液进行减压蒸馏,收集蒸馏温度180℃,真空度100pa下的馏分,得到无色透明液体,即为目标产物(8)
[0087]
对制得的产品进行如下表征:
[0088]
气相色谱分析gc:gc
‑
2014c,日本岛津公司,柱子db
‑
5,经测试,产品含量为98.7%。
[0089]
元素分析:ce
‑
440型有机碳氢氮氧硫元素分析仪,美国加联公司,经测试,元素为c:78.80%(78.79%),h:9.56%(9.55%)。
[0090]1h
‑
nmr(cdcl3为氘代试剂,四甲基硅烷为内标):6.23ppm(2h), 3.90ppm(1h),2.89ppm(1h),2.27ppm(1h),2.13ppm(1h),2.02ppm(3h),1.88ppm (1h),1.70ppm(3h),1.59ppm(2h),1.50ppm(1h),1.43ppm(4h),0.96ppm(6h)。
[0091]
实施例9
[0092]
本实施例提供制备一种环烯烃化合物其制备方法如下:
[0093]
将双环戊二烯(79.2g,0.6mol)、(232g,1.0mol)及对羟基苯甲醚(0.1556g)投于反应釜,抽真空通氮气,反复操作3次置换釜内空气,通入氮气至釜内压力
2.0mpa,开启加热及搅拌,升温至230℃,反应2h后,冷却出料,对反应结束液进行减压蒸馏,收集蒸馏温度180℃,真空度50pa下的馏分,得到无色透明液体,即为目标产物(9)
[0094]
对制得的产品进行如下表征:
[0095]
气相色谱分析gc:gc
‑
2014c,日本岛津公司,柱子db
‑
5,经测试,产品含量为98.3%。
[0096]
元素分析:ce
‑
440型有机碳氢氮氧硫元素分析仪,美国加联公司,经测试,元素为c:80.52%(80.50%),h:8.76%(8.78%)。
[0097]1h
‑
nmr(cdcl3为氘代试剂,四甲基硅烷为内标):6.23ppm(2h), 3.90ppm(1h),3.77ppm(1h),2.27ppm(2h),2.13ppm(2h),2.02ppm(3h), 1.88ppm(2h),1.70ppm(3h),1.60ppm(2h),1.50ppm(1h),1.43ppm(5h), 1.33ppm(2h)。
[0098]
实施例10
[0099]
本实施例提供制备一种环烯烃化合物其制备方法如下:
[0100]
将双环戊二烯(79.6g,0.6mol)、(246g,1.0mol)及对苯二酚(0.0326g)投于反应釜,抽真空通氮气,反复操作3次置换釜内空气,通入氮气至釜内压力1.5mpa,开启加热及搅拌,升温至230℃,反应4h后,冷却出料,对反应结束液进行减压蒸馏,收集蒸馏温度180℃,真空度30pa下的馏分,得到无色透明液体,即为目标产物(10)
[0101]
对制得的产品进行如下表征:
[0102]
气相色谱分析gc:gc
‑
2014c,日本岛津公司,柱子db
‑
5,经测试,产品含量为98.0%。
[0103]
元素分析:ce
‑
440型有机碳氢氮氧硫元素分析仪,美国加联公司,经测试,元素为c:80.73%(80.73%),h:9.05%(9.03%)。
[0104]1h
‑
nmr(cdcl3为氘代试剂,四甲基硅烷为内标):6.20ppm(2h), 3.90ppm(1h),3.77ppm(1h),2.37ppm(1h),2.27ppm(1h),2.13ppm(2h), 2.00ppm(3h),1.88ppm(2h),1.70ppm(3h),1.60ppm(1h),1.50ppm(2h), 1.40ppm(5h),1.24ppm(1h),0.96ppm(3h)。
[0105]
实施例11
[0106]
本实施例提供制备一种环烯烃化合物其制备方法如下:
[0107]
将双环戊二烯(79.2g,0.6mol)、(246g,1.0mol)及吩噻嗪(0.0065g)投于反应釜,抽真空通氮气,反复操作3次置换釜内空气,通入氮气至釜内压力1.5mpa,开启加热及搅拌,升温至230℃,反应3h后,冷却出料,对反应结束液进行减压蒸馏,收集蒸馏温度180℃,真空度30pa下的馏分,得到无色透明液体,即为目标产物(11)
[0108]
对制得的产品进行如下表征:
[0109]
气相色谱分析gc:gc
‑
2014c,日本岛津公司,柱子db
‑
5,经测试,产品含量为98.0%。
[0110]
元素分析:ce
‑
440型有机碳氢氮氧硫元素分析仪,美国加联公司,经测试,元素为c:80.73%(80.73%),h:9.05%(9.03%)。
[0111]1h
‑
nmr(cdcl3为氘代试剂,四甲基硅烷为内标):6.20ppm(2h), 3.90ppm(1h),3.77ppm(1h),2.37ppm(1h),2.27ppm(1h),2.13ppm(2h), 2.00ppm(3h),1.88ppm(2h),1.70ppm(3h),1.60ppm(1h),1.50ppm(2h), 1.40ppm(5h),1.24ppm(1h),0.96ppm(3h)。
[0112]
实施例12
[0113]
本实施例提供制备一种环烯烃化合物其制备方法如下:
[0114]
将双环戊二烯(79.2g,0.6mol)、(260g,1.0mol)及吩噻嗪(0.0678g)投于反应釜,抽真空通氮气,反复操作3次置换釜内空气,通入氮气至釜内压力1.5mpa,开启加热及搅拌,升温至230℃,反应3h后,冷却出料,对反应结束液进行减压蒸馏,收集蒸馏温度190℃,真空度50pa下的馏分,得到无色透明液体,即为目标产物(12)
[0115]
对制得的产品进行如下表征:
[0116]
气相色谱分析gc:gc
‑
2014c,日本岛津公司,柱子db
‑
5,经测试,产品含量为98.3%。
[0117]
元素分析:ce
‑
440型有机碳氢氮氧硫元素分析仪,美国加联公司,经测试,元素为c:80.95%(80.94%),h:9.25%(9.26%)。
[0118]1h
‑
nmr(cdcl3为氘代试剂,四甲基硅烷为内标):6.20ppm(2h), 3.90ppm(1h),3.77ppm(1h),2.37ppm(1h),2.27ppm(1h),2.13ppm(2h),2.00ppm(3h),1.88ppm(2h),1.70ppm(3h),1.60ppm(2h),1.50ppm(1h), 1.40ppm(5h),0.96ppm(6h)。
[0119]
实施例13
[0120]
本实施例提供制备一种环烯烃化合物其制备方法如下:
[0121]
将双环戊二烯(79.2g,0.6mol)、(246g,1.0mol)及吩噻嗪 (0.0650g)投于反应釜,抽真空通氮气,反复操作3次置换釜内空气,通入氮气至釜内压力2.0mpa,开启加热及搅拌,升温至230℃,反应4h后,冷却出料,对反应结束液进行减压蒸馏,收集蒸馏温度180℃,真空度10pa下的馏分,得到无色透明液体,即为目标产物(13)
[0122]
对制得的产品进行如下表征:
[0123]
气相色谱分析gc:gc
‑
2014c,日本岛津公司,柱子db
‑
5,经测试,产品含量为98.7%。
[0124]
元素分析:ce
‑
440型有机碳氢氮氧硫元素分析仪,美国加联公司,经测试,元素为c:80.75%(80.73%),h:9.02%(9.03%)。
[0125]1h
‑
nmr(cdcl3为氘代试剂,四甲基硅烷为内标):6.20ppm(2h), 3.90ppm(1h),2.90ppm(1h),2.27ppm(1h),2.13ppm(1h),2.00ppm(4h), 1.88ppm(1h),1.70ppm(4h),1.50ppm(3h),1.40ppm(10h)。
[0126]
实施例14
[0127]
本实施例提供制备一种环烯烃化合物其制备方法如下:
[0128]
将双环戊二烯(79.2g,0.6mol)、(260g,1.0mol)及吩噻嗪(0.0678g)投于反应釜,抽真空通氮气,反复操作3次置换釜内空气,通入氮气至釜内压力0.1mpa,开启加热及搅拌,升温至230℃,反应5h后,冷却出料,对反应结束液进行减压蒸馏,收集蒸馏温度190℃,真空度20pa下的馏分,得到无色透明液体,即为目标产物(14)
[0129]
对制得的产品进行如下表征:
[0130]
气相色谱分析gc:gc
‑
2014c,日本岛津公司,柱子db
‑
5,经测试,产品含量为98.5%。
[0131]
元素分析:ce
‑
440型有机碳氢氮氧硫元素分析仪,美国加联公司,经测试,元素为c:80.95%(80.94%),h:9.25%(9.26%)。
[0132]1h
‑
nmr(cdcl3为氘代试剂,四甲基硅烷为内标):6.20ppm(2h), 3.90ppm(1h),2.90ppm(1h),2.30ppm(1h),2.13ppm(2h),2.00ppm(3h), 1.88ppm(2h),1.70ppm(3h),1.60ppm(1h),1.50ppm(2h),1.40ppm(8h), 1.20ppm(1h),0.96ppm(3h)。
[0133]
实施例15
[0134]
本实施例提供制备一种环烯烃化合物其制备方法如下:
[0135]
将双环戊二烯(79.2g,0.6mol)、(260g,1.0mol)及吩噻嗪(0.0678g)投于反应釜,抽真空通氮气,反复操作3次置换釜内空气,通入氮气至釜内压力1.5mpa,开启加热及搅拌,升温至230℃,反应4h后,冷却出料,对反应结束液进行减压蒸馏,收集蒸馏温度190℃,真空度20pa下的馏分,得到无色透明液体,即为目标产物(15)
[0136]
对制得的产品进行如下表征:
[0137]
气相色谱分析gc:gc
‑
2014c,日本岛津公司,柱子db
‑
5,经测试,产品含量为98.3%。
[0138]
元素分析:ce
‑
440型有机碳氢氮氧硫元素分析仪,美国加联公司,经测试,元素为c:80.95%(80.94%),h:9.25%(9.26%)。
[0139]1h
‑
nmr(cdcl3为氘代试剂,四甲基硅烷为内标):6.20ppm(2h), 3.90ppm(1h),2.90ppm(1h),2.30ppm(1h),2.13ppm(2h),2.00ppm(3h), 1.88ppm(2h),1.70ppm(3h),1.60ppm(1h),1.50ppm(2h),1.40ppm(8h), 1.20ppm(1h),0.96ppm(3h)。
[0140]
实施例16
[0141]
本实施例提供制备一种环烯烃化合物其制备方法如下:
[0142]
将双环戊二烯(79.2g,0.6mol)、(274g,1.0mol)及吩噻嗪(0.0706g)投于反应釜,抽真空通氮气,反复操作3次置换釜内空气,通入氮气至釜内压力2.0mpa,开启加热及搅拌,升温至230℃,反应5h后,冷却出料,对反应结束液进行减压蒸馏,收集蒸馏温度200℃,真空度10pa下的馏分,得到无色透明液体,即为目标产物(16)
[0143]
对制得的产品进行如下表征:
[0144]
气相色谱分析gc:gc
‑
2014c,日本岛津公司,柱子db
‑
5,经测试,产品含量为98.5%。
[0145]
元素分析:ce
‑
440型有机碳氢氮氧硫元素分析仪,美国加联公司,经测试,元素为c:81.15%(81.13%),h:9.45%(9.47%)。
[0146]1h
‑
nmr(cdcl3为氘代试剂,四甲基硅烷为内标):6.20ppm(2h), 3.90ppm(1h),2.90ppm(1h),2.30ppm(1h),2.10ppm(2h),2.00ppm(3h), 1.88ppm(2h),1.70ppm(3h),1.60ppm(2h),1.50ppm(1h),1.40ppm(8h), 0.96ppm(6h)。
[0147]
性能测试:
[0148]
将实施例1
‑
16制得的环烯烃单体(1)
‑
(16)与乙烯进行加成共聚反应:
[0149]
共聚反应在2l的反应釜中进行,先用高纯氮气抽排四次,排出釜内空气。然后在氮气保护下边搅拌边依次加入甲苯900ml、实施例1
‑
16制得的环烯烃单体(1)
‑
(16)100ml、甲基铝氧烷(mao)240μmol,最后加入30μmol催化剂快速升温至80℃,并持续通入乙烯气体保持乙烯压力在 2.0mpa,反应3h后停止搅拌。聚合反应结束后,用酸化乙醇终止,沉淀析出白色粉末状固体,用乙醇洗涤抽滤,得到环烯烃共聚物a
‑
1~a
‑
16,在70℃真空干燥8h后测试。
[0150]
环烯烃共聚物(coc)结构式为:
[0151]
环烯烃摩尔含量测试方法为:用bruker amx 500核磁共振仪测定聚合物中环烯烃的摩尔含量,采用
13
c
‑
nmr,cdcl3为氘代试剂,四甲基硅烷为内标。
[0152]
玻璃化转变温度tg测试方法:使用高温凝胶渗透色谱仪(型号:agilent plgel mixed
‑
bls,生产厂家:美国安捷伦公司),流动相为1,2,4
‑
三氯苯,柱温为150℃,流速为1ml/min,进样体积为200μl,标准品为窄分布聚苯乙烯。
[0153]
测试结果如表1所示:
[0154]
表1
[0155][0156]
由表1数据可知:采用本发明提供的环烯烃化合物与乙烯加成共聚制得的环烯烃共聚物的玻璃化转变温在相同的环烯烃插入率下明显比降冰片烯或者四环十二碳烯与乙烯的共聚物高,其耐热性更佳。
[0157]
申请人声明,本发明通过上述实施例来说明本发明的一种环烯烃化合物及其制备方法和应用,但本发明并不局限于上述实施例,即不意味着本发明必须依赖上述实施例才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
[0158]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
[0159]
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1