一种制备邻甲酰胺基苯甲酰胺类化合物的方法与流程
1.本发明属于有机合成化学领域,具体涉及一种制备邻甲酰胺基苯甲酰胺类化合物的方法。
背景技术:
2.杜邦公司于2000年成功研发出邻甲酰胺基苯甲酰胺类杀虫剂,该类杀虫剂不仅对鳞翅目类害虫有较好的活性,还对特定的双翅目类害虫有较好的活性,具有广阔的应用空间和巨大的市场前景,该类杀虫剂中的代表化合物氯虫苯甲酰胺是全球销售额第一的杀虫剂。
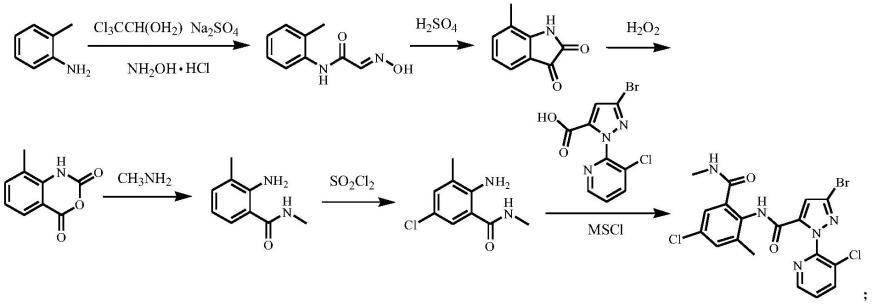
3.合成氯虫苯甲酰胺的两个中间体主要为含芳环结构的2-氨基-5-氯-3-甲基苯甲酸(中间体i)和含吡唑环结构的3-溴-1-(3-氯-2-吡啶基)-1h-吡唑-5-羧酸(中间体ii)。现有的合成方法主要有以下几种:
4.方法一,以邻氨基苯甲酸为原料,经肟化、成环、氧化、氨解、氯化五步反应得到中间体i,再与中间体 ii氨解得到氯虫苯甲酰胺。此路线起始原料便宜易得,但肟化、成环造成大量废盐废水,总收率只有26%左右,导致总成本较高。方法一具体合成路线如下式所示:
[0005][0006]
方法二,以2-硝基-3-甲基苯甲酸为原料,经h2还原、磺酰氯氯化、光气环合、一甲氨水溶液氨解得到中间体2-氨基-5-氯-n,3-二甲基苯甲酰胺,接着在甲磺酰氯、缚酸剂3-甲基吡啶作用与中间体ii进行酰胺化反应得到氯虫苯甲酰胺。此方法收率尚可,但溶剂用量多、更换频繁,后处理繁琐:
[0007]
技术实现要素:
[0008]
鉴于以上所述现有技术的缺点,本发明的目的在于提供一种原料低廉、后处理操作简便、反应条件温和的制备邻甲酰胺基苯甲酰胺类化合物的方法。
[0009]
为实现上述目的及其它相关目的,本发明提供了一种制备邻甲酰胺基苯甲酰胺类化合物的方法,具体为使式iii化合物与式iv化合物在任选取代的吡啶化合物存在的条件下反应制备得到式ii化合物、再经氨解反应得到式i化合物:
[0010][0011]
式中,r1、r2、r3、r4分别独立地表示h、卤素、cn、c
1-c6烷基、卤代c
1-c6烷基或c
1-c6烷氧基,r5表示c
1-c6烷基或c
3-c6环烷基取代的c
1-c6烷基,x表示c
1-c6烷基,y表示卤素。
[0012]
进一步地,式iii与式iv化合物反应在溶剂中进行;所述的溶剂为氯仿、二氯乙烷、乙腈、四氢呋喃、甲苯中的一种或几种。
[0013]
再进一步地,所述式iii与式iv化合物的投料摩尔比为1:1.0~1.05;
[0014]
再进一步地,所述式iv化合物与任选取代的吡啶化合物的投料摩尔比为1:1~2.3。
[0015]
再进一步地,式iii与iv化合物反应的反应温度为10~40℃;所述酰胺化反应时间为1~3h。
[0016]
进一步地,所述任选取代的吡啶化合物为无取代基的吡啶或有取代基的吡啶,所述有取代基的吡啶为被 1-3个c
1-c6烷基取代基取代的吡啶。
[0017]
进一步地,所述被1-3个c
1-c6烷基取代基取代的吡啶为2-甲基吡啶、3-甲基吡啶、4-甲基吡啶、2,6-二甲基吡啶、1,2,3-三甲基吡啶、1,2,4-三甲基吡啶、1,2,5-三甲基吡啶、2,3,4-三甲基吡啶或1,2,4-三甲基吡啶中的任一种。
[0018]
进一步地,所述氨解反应为在一甲胺的甲醇溶液中反应或在乙胺的乙醇溶液中反应。
[0019]
再进一步地,所述氨解反应的反应温度为45℃~80℃;所述的氨解反应的反应时间为1h~3h。
[0020]
再进一步地,所述式ii化合物与一甲胺或乙胺的投料摩尔比为1:1~1.3。
[0021]
进一步地,所述式iii化合物制备路线如下所示:
[0022][0023]
进一步地,当r3为cl时,所述式iii化合物是由iii’化合物与so2cl2化合物反应制备得到。
[0024]
再进一步地,以3-甲基-2-硝基苯甲酸为原料,经酰氯化反应和酯化反应,得到3-甲基-2-硝基苯甲酸甲酯,经还原反应,得到2-氨基-3-甲基苯甲酸甲酯,经氯化反应或氰化反应,得到2-氨基-5-氯-3-甲基苯甲酸甲酯。
[0025]
再进一步地,所述酰氯化反应的反应温度为50-80℃,反应时间为0.5-1h;所述的酯化反应温度在25-50℃滴加醇溶液,反应时间为1-3h。
[0026]
再进一步地,所述的溶剂为氯仿、二氯乙烷、乙腈、四氢呋喃、甲苯中的一种或几种;
[0027]
所述的酰氯化试剂为pcl3、socl2、mscl中的一种;
[0028]
所述的醇为甲醇、乙醇、异丙醇、正丁醇中的一种;
[0029]
所述的3-甲基-2-硝基苯甲酸与酰氯化试剂和醇的投料摩尔比为1:1~1.3:1~1.2。
[0030]
再进一步地,所述还原反应为2-硝基-3-甲基苯甲酸酯与氢气在催化剂的条件下在甲醇中进行,所述的催化剂为5%~10%的钯碳。
[0031]
再进一步地,所述的2-硝基-3-甲基苯甲酸酯与钯碳的投料质量比为1:0.005~0.01;氢气压力为0.5mpa~ 1.6mpa;还原反应的反应温度为50℃~100℃;所述的还原反应的反应时间为3h~9h。
[0032]
再进一步地,所述的氯化反应在溶剂中进行。
[0033]
再进一步地,所述的溶剂为氯仿、二氯乙烷、乙腈、四氢呋喃、甲苯中的一种或几种;
[0034]
所述的氯化试剂为氯气、磺酰氯、ncs中的一种或几种;
[0035]
所述氯化温度为-5℃~20℃;所述溶剂与2-氨基-5-氯-3-甲基苯甲酸酯质量为5~12倍。
[0036]
再进一步地,所述的氰化反应在溶剂中进行。
[0037]
再进一步地,所述的氰化反应溶剂为冰乙酸水溶液。
[0038]
再进一步地,所述的氰化反应步骤具体为:在40~60%的冰乙酸水溶液中,滴加40%溴氢酸,滴加完毕后升温至30℃,滴入30%双氧水,反应15min,经调碱至ph=5~6,过滤、水洗、干燥得2-氨基-5-溴-3-甲基苯甲酸酯;室温投入2-氨基-5-溴-3-甲基苯甲酸酯、n-甲基吡咯烷酮、氰化亚铜,搅拌升温回流3~5h,自然降温,加入15%氨水和3倍体积的二氯乙烷,搅拌、分液,有机层脱溶得黄色固体产物。
[0039]
进一步地,所述式iv化合物制备路线如下所示:
[0040][0041]
进一步地,所述式iv化合物是由iv’化合物与so2y2化合物反应制备得到。
[0042]
进一步地,式中,
[0043]
r1选自ch3、cl或br,
[0044]
r2为h,
[0045]
r3为cl或cn,
[0046]
r4为h、f或och3,
[0047]
r5表示ch3或
[0048]
x表示ch3或ch2ch3,
[0049]
y表示cl。
[0050]
进一步地,式中,
[0051]
r1为ch3,r2为h,r3为cl,r4为h,r5为ch3,x为ch3,y为cl;或,
[0052]
r1为ch3,r2为h,r3为cn,r4为h,r5为ch3,x为ch3,y为cl;或,
[0053]
r1为cl,r2为h,r3为cl,r4为h,r5为ch3,x为ch3,y为cl;或,
[0054]
r1为cl,r2为h,r3为cl,r4为f,r5为ch3,x为ch3,y为cl;或,
[0055]
r1为cl,r2为h,r3为cl,r4为och3,r5为ch3,x为ch3,y为cl;或,
[0056]
r1为br,r2为h,r3为cl,r4为h,r5为x为ch3,y为cl。
[0057]
由于采用了以上技术,本发明与现有技术相比,其显著优点为:
[0058]
1)本发明采用吡啶及取代物作为有机碱,相较于无机碱,起到缚酸剂作用的同时,能降低反应温度,抑制杂质生成;同时吡啶及取代物可在酸酐成环、水解以及酯化等反应中起到催化作用。
[0059]
2)本发明在胺解反应中采用甲胺的甲醇或乙醇溶液,再加入适量醇钠可加快反应速度;避免了乙腈为溶剂在高温碱性体系下易产生杂质的问题,且乙腈成本较高。
具体实施方式
[0060]
为了更好地理解本发明,以下结合具体实施案例对本发明做进一步详细说明。这些实施例是用于说明本发明的主要反应及基本特征,不受以下实施案例的限制,实施案例中采用的实施条件可以根据具体要求做进一步的调整,未注明的实施条件通常为常规实验中的条件。
[0061]
下面结合实施例,通过对实施例的描述,对本发明做进一步说明。
[0062]
实施例1
[0063]
1)2-硝基-3-甲基苯甲酸甲酯的合成:
[0064]
将36.9g2-硝基-3-甲基苯甲酸、110.8g二氯乙烷加入到四口烧瓶中,开启搅拌升
温至50℃,缓慢滴加28.6g 的socl2,0.5h滴加完毕后,升温至65℃保温0.5h,原料反应完毕后降温至40℃,开始滴加7.8g甲醇,0.5h 内滴加完毕,保温1.5h,反应完毕后负压脱溶,加158g甲醇溶解备用。
[0065]
2)2-氨基-3-甲基苯甲酸甲酯的合成:
[0066]
将步骤(1)中产物溶液转移至氢化釜中,加入0.2g 10%的钯碳后关闭氢化釜,氮气置换3次后通入0.5mpa 氢气,开启搅拌升温60℃反应5h,反应完毕后趁热过滤,滤液减压蒸馏得到2-氨基-3-甲基苯甲酸甲酯32.1g, hplc测定纯度98.7%,两步合并收率为94.2%。
[0067]
3)2-氨基-5-氯-3-甲基苯甲酸甲酯的合成:
[0068]
将30g 2-氨基-3-甲基苯甲酸甲酯投入四口瓶中,加入150g二氯乙烷,5~10℃下搅拌滴加磺酰氯,3h滴加完毕后,升温至30度保温1h,反应完毕后过滤,滤饼用30g二氯乙烷淋洗后保存备用。此步产物经干燥后得86.4g,hplc测定纯度为98.6%,收率为97.1%。
[0069]
4)3-溴-1-(3-氯-2-吡啶基)-1h-吡唑-5-酰氯的合成:
[0070]
将61.7g 3-溴-1-(3-氯-2-吡啶基)-1h-吡唑-5-羧酸、250g二氯乙烷加入到四口烧瓶中,开启搅拌升温至50℃,缓慢滴加28.6g的socl2,0.5h滴加完毕后,升温至65℃保温0.5h,原料反应完毕后负压脱除体系内残余socl2、 hcl和so2,得反应液261g保存备用。
[0071]
5)3-溴-n-[4-氯-2-甲基-6-((氧基)羰基)苯基]-1-(3-氯-2-吡啶基)-1h-吡唑-5-酰胺的合成:
[0072]
将2-氨基-5-氯-3-甲基苯甲酸甲酯湿粉投入干燥四口烧瓶中,加100g二氯乙烷分散均匀,检测水分合格后,加入35g 3-甲基吡啶,降温至10℃,缓慢滴加上步得到的含3-溴-1-(3-氯-2-吡啶基)-1h-吡唑-5-酰氯的反应液261g,0.5h滴加完毕后,升温至30℃保温1h,反应完毕后过滤、淋洗,滤饼投入下一步。
[0073]
6)氯虫苯甲酰胺的合成:
[0074]
将上步得到的3-溴-n-[4-氯-2-甲基-6-((氧基)羰基)苯基]-1-(3-氯-2-吡啶基)-1h-吡唑-5-酰胺湿粉投入四口烧瓶中,加入190g甲醇,搅拌升温至60℃并缓慢滴入20.9g30%一甲胺甲醇溶液,2h滴加完毕,60℃保温1h,反应完毕后过滤,滤饼加100g水升温打浆0.5h,趁热过滤,产物烘干后得89.9g,灰白色粉状,收率95.8%(以 3-溴-1-(3-氯-2-吡啶基)-1h-吡唑-5-酰氯计),hplc(高效液相色谱)测定纯度98.8%。
[0075]
实施例2
[0076]
1)2-氨基-5-溴-3-甲基苯甲酸甲酯的合成:
[0077]
将30g 2-氨基-3-甲基苯甲酸甲酯投入四口瓶中,加入100g50%冰乙酸,室温下搅拌滴加40g 40%的氢溴酸, 0.5h滴加完毕后,升温至30℃,缓慢滴入12.5g 30%h2o2,15min滴加完毕,保温0.5h至反应完毕,加入1.5g 亚硫酸氢钠,naoh调节体系ph至5~6,降温抽滤,干燥得产物42.8g,hplc测定纯度为97.9%,收率为94.5%。
[0078]
2)2-氨基-5-氰-3-甲基苯甲酸甲酯的合成:
[0079]
取上步得到的2-氨基-5-溴-3-甲基苯甲酸甲酯、200g n-甲基吡咯烷酮、16.2g氰化亚铜投入反应瓶中,升温至回流并保温3~5h,反应完毕后加入120g 15%的氨水和140g二氯乙烷,搅拌10min后静置分液,有机层脱溶得黄色固体31.8g,hplc测定纯度为98.1%,计得收率95.5%。
[0080]
3)3-溴-n-[4-氰基-2-甲基-6-((氧基)羰基)苯基]-1-(3-氯-2-吡啶基)-1h-吡唑-5-酰胺的合成:
[0081]
取13g 2-氨基-5-氰基-3-甲基苯甲酸甲酯投入干燥四口烧瓶中,加120g二氯乙烷分散均匀,检测水分合格后,加入7.4g 3-甲基吡啶,降温至10℃,缓慢滴加52.5g 40%质量分数的3-溴-1-(3-氯-2-吡啶基)-1h-吡唑-5
‑ꢀ
酰氯二氯乙烷溶液,0.5h滴加完毕后,升温至45℃保温1h,反应完毕后过滤、淋洗,滤饼投入下一步。
[0082]
4)溴氰虫酰胺的合成:
[0083]
将3-溴-n-[4-氰基-2-甲基-6-((氧基)羰基)苯基]-1-(3-氯-2-吡啶基)-1h-吡唑-5-酰胺湿粉投入四口烧瓶中,加入120g甲醇,搅拌升温至60℃并缓慢滴入9.5g 30%一甲胺甲醇溶液,2h滴加完毕,60℃保温1h,反应完毕后过滤,产物烘干后得白色粉末30.1g,收率95.4%(以3-溴-1-(3-氯-2-吡啶基)-1h-吡唑-5-酰氯计),hplc(高效液相色谱)测定纯度98.2%。
[0084]
上述实施例仅为本发明的优选技术方案,而不应视为对于本发明的限制,本发明的保护范围应以权利要求记载的技术方案,包括权利要求记载的技术方案中技术特征的等同替换方案为保护范围,即在此范围内的等同替换改进,也在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1