一种高活性物含量N-酰基甲基牛磺酸钠的连续化制备方法与流程
一种高活性物含量n-酰基甲基牛磺酸钠的连续化制备方法
技术领域
1.本发明属于日化材料技术领域,具体涉及一种高活性物含量n-酰基甲基牛磺酸钠的连续化制备方法。
背景技术:
2.目前工业化生产n-酰基甲基牛磺酸钠多采用以酰氯为主要原料的肖顿鲍曼缩合工艺:脂肪酰氯与甲基牛磺酸钠在碱性条件下,在水/有机溶剂中进行酰胺化反应。如专利cn200510023559.7公开了一种高纯度n-酰基-n-甲基牛磺酸钠的合成方法,系在水和亲水性混合溶媒中使n-甲基牛磺酸钠和脂肪酰氯于无机碱存在下反应制备高纯度的n-酰基-n-甲基牛磺酸钠。专利cn201210403227.1公开了一种n-酰基-n-甲基牛磺酸的合成方法:以牛磺酸钠和脂肪酰基氯在水和丙酮混合溶剂中加碱反应,得n-酰基牛磺酸钠;然后再加入氢氧化钠,滴加硫酸二甲酯继续反应,得n-酰基-n-甲基牛磺酸。专利cn201910417964.9公开了一种n-脂肪酰基-n-甲基牛磺酸钠的制备方法,包括:以水为溶剂,n-甲基牛酸酸钠与脂肪酰氯反应生成n-脂肪酰基-n-甲基牛磺酸钠,调节ph为碱性,然后与水混溶的有机溶剂混合;再调节ph值并置于结晶温度使析出n-脂肪酰基-n-甲基牛磺酸钠结晶。
3.上述工艺路线以酰氯为原料,引入了较多氯离子,后续脱盐步骤会产生大量含氯废水,环保性差,同时酰氯属于危险物料,安全性差,但由于该工艺反应效率极高,可达95%以上,因此成为主流的工业化生产工艺。同时也有部分研究人员尝试了以脂肪酸为主要原料的直接酰胺化缩合工艺,即脂肪酸和甲基牛磺酸钠在高温条件下脱水缩合一步合成目标产品。该工艺反应简单,原子经济性高,且原料安全无害,但由于其反应效率偏低,且产品中未反应的脂肪酸残留偏高,导致工业化困难。例如专利us 5496959/us 2880219/jp2002-234868/us3232968等描述脂肪酸和甲基牛磺酸钠的直接缩合工艺,分别对催化剂,反应温度,投料比等参数进行筛选优化,所得产品收率仅为50~80%,剩余的脂肪酸高达20~50%。由于此工艺所得产品中脂肪酸残留较高,因此有较多的研究者通过溶剂洗涤的方式去除脂肪酸,进而获得活性物含量较高的产品,cn105175291/cn201510568940.5等研究者对此做了详细研究。通过脂肪酸和甲基牛磺酸钠直接酰胺化缩合工艺获得高活性物含量n-酰基甲基牛磺酸钠的连续化制备方法还未见报道。
技术实现要素:
4.针对以上现有技术存在的缺点和不足之处,本发明的目的在于提供一种高活性物含量n-酰基甲基牛磺酸钠的连续化制备方法。本发明方法以脂肪酸为原料,在无需使用催化剂的情况下,通过特定的混合溶剂可实现目标产品n-酰基甲基牛磺酸钠和脂肪酸的快速分离,并实现连续化生产,既得到了高活性物的目标产品,同时有效的提高了生产效率。
5.本发明目的通过以下技术方案实现:
6.一种高活性物含量n-酰基甲基牛磺酸钠的连续化制备方法,包括如下制备步骤:
7.(1)将脂肪酸和甲基牛磺酸钠泵入管式反应器中,控制管式反应器内温度为150~
250℃进行反应;
8.(2)步骤(1)管式反应器内反应后的物料进入静态混合器与混合溶剂进行预混合,混合液通过连续分液塔进行连续分液;所述混合溶剂为水和有机溶剂的混合;
9.(3)步骤(2)连续分液后的重组分经喷粉干燥得到高活性物含量n-酰基甲基牛磺酸钠,轻组分进行溶剂回收后重复使用。
10.进一步地,步骤(1)中所述脂肪酸先加热至50~80℃熔融预处理后泵入管式反应器中。具体为依据不同碳链脂肪酸的熔点选择不同熔融温度。
11.进一步地,步骤(1)中所述甲基牛磺酸钠先在60~120℃温度及-0.1~-0.05mpa压力下脱水预处理至固含量90%以上后泵入管式反应器中。脱水过程中需维持温度保持甲基牛磺酸钠的流动性。
12.进一步地,步骤(1)中所述脂肪酸选自碳原子数为6~22的脂肪酸。所述脂肪酸中的疏水长链为直链或具有支链的疏水长链,所述疏水长链为饱和烷基或具有一个或多个不饱和基团的烷基。优选地,所述脂肪酸可选择为油酸、亚油酸、亚麻酸、辛酸、癸酸、月桂酸、肉豆蔻酸、软脂酸、硬脂酸、椰油酸中的至少一种,更优选为月桂酸和椰油酸中的至少一种。
13.进一步地,步骤(1)中所述管式反应器的反应温度优选150~210℃,更优选160~200℃。反应温度超过200℃产品有黄变的风险,反应温度低于160℃产品转化率偏低。
14.进一步地,步骤(1)中所述脂肪酸和甲基牛磺酸钠均通过输料泵泵入管式反应器中,输料泵进行伴热处理,脂肪酸的进料速度为0.1~100l/min,优选25~100l/min,更优选为50~100l/min;甲基牛磺酸钠的进料速度为0.1~100l/min,优选0.1~60l/min,更优选为10~40l/min。脂肪酸进料速度低于50l/min或甲基牛磺酸钠的进料速度高于40l/min,可能会导致甲基牛磺酸钠局部浓度偏高而使产品粘度偏高堵塞管道,因为脂肪酸在合成过程中须保持过量充当反应溶剂;脂肪酸进料速度高于100l/min或甲基牛磺酸钠的进料速度低于10l/min时,可能会导致甲基牛磺酸钠局部浓度偏低而使产品残留脂肪酸偏高,反应经济性较差。
15.进一步地,步骤(1)中所述管式反应器内反应停留时间为5~20min,优选5~15min,最优选6~10min。反应停留时间高于10min会使生产效率降低,而反应停留时间低于6min时会使得产品转化率偏低。
16.进一步地,步骤(2)中所述静态混合器的物料进口温度为100~250℃,优选100~200℃,最优选120~160℃;温度低于120℃时,产品易凝固,温度高于160℃时,与溶剂混合时放热明显导致溶剂汽化,静态混合器压力过高,导致安全风险高;混合液出口温度为25~50℃,温度高于50℃时溶剂易挥发。静态混合器辅以冷冻水降温。
17.进一步地,步骤(2)中所述有机溶剂为醇类(如甲醇、乙醇和正丙醇等醇类)、酮类(如丙酮、丁酮和戊酮等酮类)、酯类(乙酸乙酯、乙酸丙酯、乙酸异丙酯、乙酸丁酯和丙酸丁酯等酯类)、矿物油烷烃类(石蜡油和白油等矿物油烷烃类)中的至少一种溶剂,优选醇类、酯类或矿物油烷烃类有机溶剂,最优选为酯类有机溶剂;酯类有机溶剂中优选总碳数低于6的酯,更优选总碳数为5或以内的酯,如乙酸乙酯、乙酸丙酯、乙酸异丙酯、乙酸丁酯中的任意一种,最优选总碳数为4或以内的酯,如乙酸乙酯。在本发明中酯类溶剂和水的混合溶剂可使产品溶液迅速分液,分液界面清晰明显。而其它混合溶剂存在分液缓慢,静置时间偏长或使物料乳化致分层不明显的情况,因此酯类溶剂作为最优溶剂能有效解决上述问题。
18.进一步地,步骤(2)中所述混合溶剂中有机溶剂的质量分数为20~80%,优选20~50%,最优选30~50%。
19.进一步地,步骤(2)中所述混合溶剂的进料速度为50~300l/min,优选80~250l/min,最优选100~200l/min;进料速度高于200l/min时,溶剂量偏大,后续溶剂回收经济性差,进料速度低于100l/min时,溶剂量偏小,导致后续分层效果不佳或分层不明显,影响产品活性物含量。
20.进一步地,步骤(2)中经静态混合器混合降温后的物料经连续分液塔进行连续分液,连续分液塔内的温度控制在30~50℃。连续分液塔的构造图如附图1所示(参考我司专利技术cn 106345387 b),其左端与静态混合器的出口相连,下端与产品喷粉塔的进口相连,上端与溶剂精馏塔的进口相连。
21.进一步地,步骤(3)中所述喷粉干燥的温度为100~200℃,优选100~150℃,最优选120~140℃,经喷粉干燥所得产品的固含量95%以上,活性物(n-酰基甲基牛磺酸钠)含量90%以上。
22.与现有技术相比,本发明的有益效果是:
23.(1)本发明采用管式反应器连续制备n-酰基甲基牛磺酸钠,反应过程中无需使用催化剂,后处理通过混合溶剂溶解后进行连续分液,收集重组分进行干燥,所得最终产品的活性物高达90%以上。
24.(2)本发明采用脂肪酸工艺合成n-酰基甲基牛磺酸钠,成本较酰氯工艺的产品优势明显,且有效避免了酰氯工艺引入的大量氯离子;此外,在后处理步骤选择乙酸乙酯和水的混合溶剂,可快速有效地分液除去产品中过量的脂肪酸,无需使用催化剂即可得到高活性物含量(90%以上)的产品n-酰基甲基牛磺酸钠,且能实现产品的连续化生产。
附图说明
25.图1为本发明连续化制备方法所使用到的连续分液塔的构造图;
26.图2为本发明实施例中连续化制备方法的工艺流程图。
具体实施方式
27.下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
28.实施例1
29.本实施例的一种甲基月桂酰基牛磺酸钠的连续化制备方法,其工艺流程图如图2所示,具体制备步骤如下:
30.(1)将原料甲基牛磺酸钠和月桂酸经过预处理,月桂酸在熔酸釜中进行熔化,熔酸釜温度为60℃,釜内保持氮气氛围,搅拌熔化至流动液体状态保温待用;甲基牛磺酸钠在脱水釜中进行真空脱水,脱水压力-0.095mpa,脱水温度从70℃不断升高以保证釜内液体状态,终点温度110℃,取样测得固含量97.6%。
31.(2)将步骤(1)预处理后的甲基牛磺酸钠和月桂酸通过输送泵泵入管道反应器,甲基牛磺酸钠流速为20l/min,月桂酸流速为79l/min,停留时间为6min,管道反应器温度为195℃,管道反应器出口取样,固含量98.1%,活性物含量48.2%,月桂酸含量47.1%。
32.(3)步骤(2)反应完物料进入静态混合器与混合溶剂混合,混合溶剂为水和乙酸乙酯的混合物,乙酸乙酯的质量分数为40%,进口温度为140℃,进料流量为180l/min,静态混合器辅以冷冻水降温,静态混合器出口温度为40℃,静态混合器出口与连续分液塔相连,分液塔辅以伴热,保持温度40℃,经连续分液塔分液后,轻组分中的乙酸乙酯通过减压蒸馏回收套用,重组分直接进喷粉塔干燥,干燥所得产品固含量98.7%,活性物含量94.5%。
33.实施例2
34.本实施例的一种甲基椰油酰基牛磺酸钠的连续化制备方法,其工艺流程图如图2所示,具体制备步骤如下:
35.(1)将原料甲基牛磺酸钠和椰油酸经过预处理,月桂酸在熔酸釜中进行熔化,熔酸釜温度为70℃,釜内保持氮气氛围,搅拌熔化至流动液体状态保温待用;甲基牛磺酸钠在脱水釜中进行真空脱水,脱水压力-0.095mpa,脱水温度从70℃不断升高以保证釜内液体状态,终点温度110℃,取样测得固含量93.2%。
36.(2)将步骤(1)预处理后的甲基牛磺酸钠和椰油酸通过输送泵泵入管道反应器,甲基牛磺酸钠流速为15l/min,月桂酸流速为58l/min,停留时间为8min,管道反应器温度为200℃,管道反应器出口取样,固含量97.8%,活性物含量46.6%,月桂酸含量49.2%。
37.(3)步骤(2)反应完物料进入静态混合器与混合溶剂混合,混合溶剂为水和乙酸乙酯的混合物,乙酸乙酯的质量分数为45%,进口温度为150℃,进料流量为144l/min,静态混合器辅以冷冻水降温,静态混合器出口温度为40℃,静态混合器出口与连续分液塔相连,分液塔辅以伴热,保持温度40℃,经连续分液塔分液后,轻组分中的乙酸乙酯通过减压蒸馏回收套用,重组分直接进喷粉塔干燥,干燥所得产品固含量99.1%,活性物含量94.3%。
38.对比例1~5
39.对比例1~5与实施例1相比,静态混合器中采用不同的混合溶剂进行分液,其余完全相同,具体见下表1所示。
40.表1
[0041] 静态混合器所用溶剂有机溶剂占比分层所需时间分液温度分液状态实施例1水和乙酸乙酯40%2min40℃界面清晰对比例1纯水0乳化,无法分层//对比例2水和乙醇40%21min45℃界面模糊对比例3水和环己烷40%>30min50℃界面很模糊对比例4水和液体石蜡40%27min45℃界面模糊对比例5水和丙酮40%半乳化,无法分层//
[0042]
由表1结果可见,本发明连续化制备n-酰基甲基牛磺酸钠的方法中,分液采用的混合溶剂对合成产品质量和合成效率非常关键,当采用水和乙酸乙酯的混合溶剂相比其它混合溶剂,产物连续化分离效率和分离质量显著提高。
[0043]
对比例6~10
[0044]
对比例6~10与实施例1相比,静态混合器中采用不同比例水和乙酸乙酯的混合溶剂进行分液,其余条件完全相同,具体见下表2所示。
[0045]
表2
[0046] 混合溶剂乙酸乙酯占比分层所需时间分液温度分液状态
实施例1水和乙酸乙酯40%2min40℃界面清晰对比例6水和乙酸乙酯0乳化,无法分层//对比例7水和乙酸乙酯20%13min40℃界面模糊对比例8水和乙酸乙酯60%<2min40℃界面清晰对比例9水和乙酸乙酯80%<2min40℃底部少量产品析出对比例10水和乙酸乙酯100%乳状液,底部产品析出/底部大量产品析出
[0047]
由表2结果可见,采用水和乙酸乙酯的混合溶剂进行连续分液时,有机溶剂占比偏低(低于40%),物料易乳化,溶液无法分层或分层界面不清晰,分液时间偏长;有机溶剂占比过高(高于40%)时,虽然不影响分液效果,但是有较多的产品会溶解于溶剂乙酸乙酯中,造成后续溶剂回收困难,且经济性较差,此外有机溶剂占比过高时,产品在水中处于过饱和状态,底部会有产品析出。综合选择有机溶剂占比40%时,产物连续化分离效率和分离质量显著提高。
[0048]
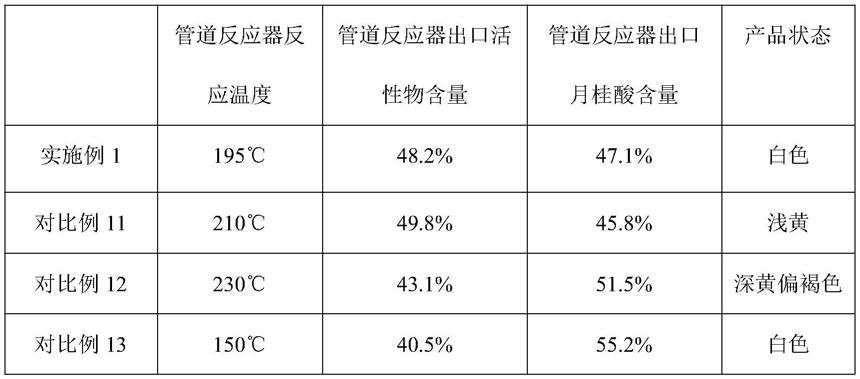
对比例11~13
[0049]
对比例11~13与实施例1相比,管道反应器中采用不同的反应温度,其余完全相同,具体见下表3所示。
[0050]
表3
[0051][0052]
由表3结果可见,本发明连续化制备n-酰基甲基牛磺酸钠的方法中,管道反应器中反应温度超过200℃产品有黄变的风险,反应温度低于160℃产品转化率偏低。
[0053]
通过上述的实施例和对比例的测试可以发现,本发明连续化制备n-酰基甲基牛磺酸钠的方法,反应过程中无需使用催化剂,并通过连续化工艺可得到活性物含量高达90%以上的产品,且在合成中避免了使用酰氯类物质,环境友好。
[0054]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其它的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1