一种从泡桐叶中提取毛蕊花糖苷的方法与流程
1.本发明属于中药技术领域,具体涉及一种从泡桐叶中提取毛蕊花糖苷的方法。
背景技术:
2.泡桐属(paulownia)植物共7种,均产自我国,除东北北部、内蒙古、新疆北部和西藏等地区外,全国均有分布、栽培或野生,有些地区也正在引种。泡桐作为一种优质的速生木材,其除了广泛地应用于农业生产外,还是一种常用的中药材,泡桐的花、叶、皮、根和果均可入药。本草纲目对泡桐各个部位的药理作用做了详细记载,现代医学研究也表明泡桐具有抑菌、消炎、抗肿瘤甚至杀虫的作用。泡桐树枝繁叶茂,具有很多的叶片,是一笔丰富的自然资源。另外,泡桐叶还可以作为饲料,不仅能够促进动物生长,而且能够提高动物的抗病能力,具有丰富的经济价值和社会价值。
3.毛蕊花糖苷又名麦角甾苷、毛蕊花苷或类叶升麻苷,是4,5-二羟基苯乙醇(羟基酪醇)通过酯键和糖苷键与以c3位糖苷键结合鼠李糖c1位β-d-吡喃葡萄糖的苯乙醇苷类化合物,其英文名为acteoside、verbascoside或kusaginin,其分子式为c
29h36o15
,分子量为624.59。植物化学研究表明,毛蕊花糖苷广泛存在于列当科、玄参科、木兰科和唇形科等多种双子叶植物中。现代药理学研究发现,毛蕊花糖苷具有显著的抗氧化、抗炎、增强记忆力、保护神经、抗肿瘤等多种药理活性,由于其来源广泛、生物活性强,且不良反应小,受到国内外研究者的广泛关注。
4.随着国内外毛蕊花糖苷等苯丙素苷类化合物药学研究的逐渐深入,其药用价值日益得到重视,市场上毛蕊花糖苷及其相关产品已供不应求。由于毛蕊花糖苷为肉苁蓉属植物的主要活性成分,近年来的研究主要集中于从肉苁蓉属植物中提取分离毛蕊花糖苷。也有报道,可以从桂花、裸花紫珠叶、凌霄属植物、密蒙花等其它植物中提取分离毛蕊花糖苷。然而,仅从这几种植物中提取制备毛蕊花糖苷很难满足实际需要,且从上述植物中分离纯化毛蕊花糖苷的方法往往需要采用反复柱层析的方法来获取高纯度的毛蕊花糖苷样品,存在耗时长和溶剂耗费量大的缺点。另外,这些植物的生物量小,提取制备需要大量种植,经济性不高。
技术实现要素:
5.有鉴于此,本发明的目的在于提供一种从泡桐叶中提取毛蕊花糖苷的方法。所述泡桐叶含量丰富,从其中提取毛蕊花糖苷无需提前大量种植,具有很好的经济价值,且提取的毛蕊花糖苷纯度高,能够达到98%以上,可以满足实际需要。
6.第一方面,本发明提供一种从泡桐叶中提取毛蕊花糖苷的方法,包括如下步骤:
7.(1)对泡桐叶进行提取,得到粗提物;
8.(2)采用大孔吸附树脂对步骤(1)得到的粗提物进行分离纯化,得到毛蕊花糖苷富集部位;
9.(3)对步骤(2)得到的毛蕊花糖苷富集部位进行液相色谱分离,得到毛蕊花糖苷收
集液后进行浓缩和干燥,得到毛蕊花糖苷;
10.所述液相色谱分离中,色谱柱填料为十八烷基键合硅胶、八烷基键合硅胶、四烷基键合硅胶或苯基键合硅胶中的任意一种;
11.所述液相色谱分离中,流动相为20~50vol%的甲醇或10~40vol%乙腈溶液。
12.优选地,步骤(1)具体为:采用选自水、甲醇或乙醇中的一种或多种为溶剂对泡桐叶进行提取,得到粗提物。
13.优选地,所述溶剂为30~50vol%的乙醇溶液。
14.优选地,所述对泡桐叶进行提取中,泡桐叶的质量与溶剂的体积的比例为1:(20~50)g/ml。
15.优选地,步骤(1)中所述提取的温度不超过70℃,更优选为40~70℃。
16.优选地,所述大孔吸附树脂包括ab-8大孔吸附树脂、dm-301大孔吸附树脂、da-201大孔吸附树脂、d-101大孔吸附树脂、hpd100大孔吸附树脂或dm-130大孔吸附树脂中的任意一种,更优选为d-101大孔吸附树脂或ab-8大孔吸附树脂,最优选为d-101大孔吸附树脂。
17.优选地,采用大孔吸附树脂对步骤(1)得到的粗提物进行分离纯化时,洗脱程序如下表所示:
18.洗脱程序
19.洗脱液洗脱体积洗脱液流速水2~4个柱体积1~2个柱体积/小时10vol%乙醇2~4个柱体积1~2个柱体积/小时30vol%乙醇2~4个柱体积1~2个柱体积/小时50vol%乙醇3~5个柱体积1~2个柱体积/小时
20.优选地,所述液相色谱分离中,流动相为30~35vol%的甲醇或20~25vol%的乙腈溶液。
21.优选地,所述液相色谱分离中,流动相的流速为2-15ml/min。
22.优选地,所述液相色谱分离中,检测波长为240~370nm。
23.与现有技术相比,本发明的有益效果为:
24.(1)本发明在白花泡桐叶的hplc谱图中发现分离度和相对含量均非常高的成分,在鉴定为毛蕊花糖苷的基础上,提供了一种从泡桐叶中提取毛蕊花糖苷的方法,填补了该领域的技术空白,扩宽了毛蕊花糖苷的植物来源,具有重大的经济价值;
25.(2)本发明提供的提取毛蕊花糖苷的方法以泡桐叶为原料,依次通过粗提取、大孔树脂吸附分离和液相色谱分离,得到的毛蕊花糖苷纯度在98%以上,与现有纯化方法相比,本纯化方法采用高效液相色谱等度洗脱,制备时间短,分离效率高,工艺简单,经济成本低,易于实现产业化和工业化。
附图说明
26.图1为毛蕊花糖苷的结构式示意图;
27.图2为白花泡桐叶的hplc指纹图谱;
28.图3为白花泡桐叶的hplc指纹图谱中11号峰化合物的核磁共振h谱图;
29.图4为白花泡桐叶的hplc指纹图谱中11号峰化合物的核磁共振c谱图;
30.图5为白花泡桐叶的hplc指纹图谱不同色谱柱考察结果图;
31.图6为白花泡桐叶的hplc指纹图谱不同溶剂系统考察结果图;
32.图7为白花泡桐叶的hplc指纹图谱不同检测波长考察结果图;
33.图8为白花泡桐叶的hplc指纹图谱不同柱温考察结果图;
34.图9为实施例4得到的毛蕊花糖苷的纯度检测色谱图。
具体实施方式
35.下面将结合本发明实施例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
36.本发明对所有实验原料的来源没有特殊的限制,从市场上购买或者按照本领域技术人员熟知的常规制备方法制备得到的即可。
37.本发明是建立在对泡桐叶提取液的色谱条件和指纹谱图大量研究的基础上做出的,在指纹谱图中首次发现了分离度和相对含量均非常高的成分,经制备分离后鉴定为毛蕊花糖苷(其结构式如图1所示),具体过程如下:
38.称取白花泡桐叶粉末0.5g,置于具塞锥形瓶中,精密加入70vol%甲醇10ml,密塞,称定质量,超声处理(功率500w,频率40khz)30min,放冷,用70vol%甲醇补足减失的质量,取上清液用0.45μm微孔滤膜滤过,制得白花泡桐叶样品溶液。
39.对白花泡桐叶样品溶液进行hplc检测并记录其指纹图谱,色谱条件为:色谱柱:agilent zorbax hc-c18(2)(4.6mm
×
250mm,5μm);流动相:甲醇(b)-0.1vol%甲酸缓冲溶液(a);检测波长:254nm;流速:1ml/min;柱温:30℃;进样量:10μl;梯度洗脱,具体洗脱程序如下表所示:
40.梯度洗脱程序
[0041][0042][0043]
白花泡桐叶样品的hplc指纹图谱如图2所示,其中图谱呈现23个特征峰,其中11号峰为共有峰,强度较高,保留时间相对居中与其余色谱峰的分离度良好,因此选定11号峰为参照峰。因不确定该峰所代表的化合物,对其进行快速制备及鉴定。
[0044]
取白花泡桐叶样品溶液,经半制备液相色谱得到11号峰化合物,色谱条件为:色谱柱ymc-pack ph(10mm
×
250mm,5μm);等度洗脱;流动相:甲醇(b)-水(a)=(33:67,体积比)。检测波长254nm;流动相流速2.5ml/min;柱温30℃;tr=24.9min。采用核磁共振光谱仪对11号峰化合物进行定性,结果如图3-4所示,其中图3为11号峰化合物的核磁共振h谱图,图4为11号峰化合物的核磁共振c谱图,数据如下所示:esi-ms m/z:647.1940[m+na]
+
。1h nmr(600mhz,meod)δ:7.59(1h,d,j=16.2hz,h-7'),7.05(1h,d,j=1.8hz,h-2'),6.95(1h,dd,j=7.8,1.8hz,h-6'),6.77(1h,d,j=7.8hz,h-5'),6.69(1h,d,j=2.4hz,h-2),6.67(1h,d,j=7.8hz,h-5),6.56(1h,dd,j=8.0,1.8hz,h-6),6.27(1h,d,j=15.6hz,h-8'),5.18(1h,d,j=1.8hz,h-1
″
'),4.37(1h,d,j=7.8hz,h-1
″
),4.05(1h,m,h-8β),3.71(1h,m,h-8α),2.79(2h,m,h-7),1.09(3h,d,j=6.0hz,h-6
″
').
13
c-nmr(meod,125mhz)δ:166.9(c-9'),148.5(c-4'),146.7(c-7'),145.5(c-3'),144.8(c-3),143.3(c-4),130.0(c-1),126.2(c-1'),121.9(c-6'),119.8(c-6),115.7(c-2),115.1(c-5'),114.9(c-5),113.7(c-8'),113.2(c-2'),102.8(c-1
″
),101.7(c-1
″
'),80.3(c-3
″
),74.8(c-2
″
),74.6(c-5
″
),72.4(c-4
″
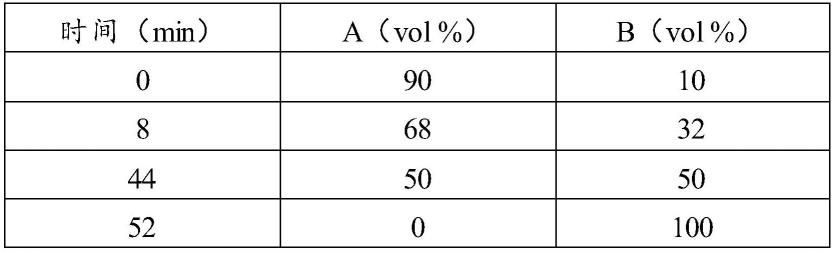
'),71.0(c-2
″
'),70.9(c-3
″
'),70.6(c-5
″
'),69.1(c-4
″
),69.0(c-5
″
'),60.9(c-6
″
),35.2(c-7),17.0(c-6
″
')。以上数据与文献报道基本一致(wang xq,et al.phenylethaniod glycosides from orobanchepycnostachya hance and their chemotaxonomic significance[j].biochemical systematics andecology.),因此最终核磁定性鉴定11号峰化合物为毛蕊花糖苷。
[0045]
基于上述研究,本发明提供一种从泡桐叶中提取毛蕊花糖苷的方法,包括如下步骤:
[0046]
(1)对泡桐叶进行提取,得到粗提物;
[0047]
(2)采用大孔吸附树脂对步骤(1)得到的粗提物进行分离纯化,得到毛蕊花糖苷富集部位;
[0048]
(3)对步骤(2)得到的毛蕊花糖苷富集部位进行液相色谱分离,得到毛蕊花糖苷收集液后进行浓缩和干燥,得到毛蕊花糖苷;
[0049]
所述液相色谱分离中,色谱柱填料为十八烷基键合硅胶、八烷基键合硅胶、四烷基键合硅胶或苯基键合硅胶中的任意一种;
[0050]
所述液相色谱分离中,流动相为20~50vol%的甲醇或10~40vol%乙腈溶液。
[0051]
在本发明中,优选采用水、甲醇或乙醇中的一种或多种为溶剂,更优选采用30~50vol%的乙醇溶液,最优选采用50vol%的乙醇溶液对泡桐叶进行提取,得到粗提物。所述提取的方式优选包括浸渍、煎煮、渗滤、回流提取、超声提取或超临界提取中的任意一种或多种,更优选包括超声提取和/或回流提取,最优选为超声提取。在本发明中,对泡桐叶进行提取时,泡桐叶的质量与溶剂的体积的比例优选为1:(20~50)g/ml,更优选为1:(20~30)g/ml,最优选为1:30g/ml。因为毛蕊花糖苷在高温下易发生转化,因此所述提取的温度优选不超过70℃,更优选为40~70℃,所述提取的时间优选为0.5~2.5h,更优选为1~1.5h,最优选为1h。
[0052]
按照本发明,在得到粗提物后采用大孔吸附树脂对粗提物进行分离纯化,得到毛蕊花糖苷富集部位。在本发明中,所述大孔吸附树脂优选包括ab-8大孔吸附树脂、dm-301大孔吸附树脂、da-201大孔吸附树脂、d-101大孔吸附树脂、hpd100大孔吸附树脂或dm-130大
孔吸附树脂中的任意一种,更优选为ab-8大孔吸附树脂或d-101大孔吸附树脂,最优选为d-101大孔吸附树脂。提高上样质量浓度有利于提高树脂的使用效率。然而,当上样质量浓度过高时,在一定体积流量下,样品与树脂的接触时间变短,同时上样液质量浓度过高,则溶液过于黏稠,会在树脂颗粒表面形成黏性吸附,严重影响吸附效果。因此,上样液质量浓度优选在15~25mg/ml,更优选为25mg/ml。
[0053]
在本发明中,50vol%的乙醇作为洗脱剂是更优选择。但因为大孔吸附树脂柱中存在杂质,优选采用水、10vol%乙醇、30vol%乙醇进行除杂后,再用50vol%的乙醇去进行洗脱。具体的洗脱程序优选如下表所示:
[0054]
洗脱程序
[0055]
洗脱液洗脱体积洗脱液流速水2~4个柱体积1~2个柱体积/小时10vol%乙醇2~4个柱体积1~2个柱体积/小时30vol%乙醇2~4个柱体积1~2个柱体积/小时50vol%乙醇3~5个柱体积1~2个柱体积/小时
[0056]
按照本发明,对毛蕊花糖苷富集部位进行液相色谱分离,得到毛蕊花糖苷收集液。在本发明中,所述液相色谱优选为制备液相色谱。所述液相色谱分离中,色谱柱填料优选为十八烷基键合硅胶、八烷基键合硅胶、四烷基键合硅胶或苯基键合硅胶中的任意一种。所述色谱柱的内径优选为10~50mm,更优选为10~30mm。所述流动相优选为20~50vol%的甲醇溶液或10~40vol%乙腈溶液,更优选为30~40vol%的甲醇或20~25vol%乙腈溶液,最优选为30~35vol%的甲醇溶液。所述流动相的流速优选为2~15ml/min,更优选为5~15ml/min。所述液相色谱分离中,检测波长优选为240~370nm,更优选为280~370nm。
[0057]
在某个具体的实施例中,液相色谱的条件优选为:
[0058]
色谱柱:ymc-pack ods-a(20mm
×
250mm,5μm);
[0059]
流动相:甲醇(b)-水(a)=(33:67,体积比),等度洗脱;
[0060]
检测波长334nm;
[0061]
流动相流速10ml/min;
[0062]
柱温30℃;
[0063]
tr=25.0min;
[0064]
进样量:20μl。
[0065]
按照本发明,在得到毛蕊花糖苷收集液后进行浓缩和干燥,得到毛蕊花糖苷固体。在本发明中,浓缩的形式没有特殊的限制,优选在不超过70℃的条件下进行真空浓缩。所述浓缩优选至固液质量体积比为1:(4~5)g/ml时结束。本发明对干燥的形式没有特殊的限制,优选采用冷冻干燥、喷雾干燥或微波干燥中的任意一种或多种,最终得到毛蕊花糖苷固体。
[0066]
本发明以泡桐叶为原料,通过常规的初提取,粗提物中毛蕊花糖苷质量分数不低于5%(最高可达11.1%),采用大孔吸附树脂进一步分离后,得到的毛蕊花糖苷富集部位的纯度高达27%以上,通过液相色谱分离后,纯度可达到98%以上,工艺简单。此外,该方法毛蕊花糖苷产品得率高(5.5%),成本低,适于工业化应用。
[0067]
需要说明的是,虽然有学者从白花泡桐花中发现了含量很低的毛蕊花糖苷(冯卫
生,吕锦锦,张靖柯,李孟,张贝贝,郑晓珂.泡桐花中糖苷类成分及其抗氧化活性.中成药.2020,42(02):369-374),然而这种研究对首次在白花泡桐叶的hplc谱图中发现非常适合做定量分析的成分,并鉴定为毛蕊花糖苷并无太多的借鉴作用,原因有二:
[0068]
一是毛蕊花糖苷这种成分来源于不同的器官,白花泡桐的花与叶是不同的器官,不同的器官生理功能有差异、对环境的响应有差异,且次生代谢产物的合成也各有侧重,从而造成其化学成分的组成差异,花中发现了较低含量的毛蕊花糖苷并不代表叶中就含有大量的毛蕊花糖苷,相反叶中可能含量更低甚至常规的分析方法根本检测不到;二是毛蕊花糖苷的获取方法有本质区别,文献报道的白花泡桐花中毛蕊花糖苷为系统分离纯化的方法获得,具有较大的偶然性,而本发明是通过指纹图谱找到共有的、含量大的一个色谱峰并对其进行定向分离制备鉴定而获得,这个共有的且含量大的色谱峰是什么化学成分是无法预测的。
[0069]
因此,本发明首次发现白花泡桐叶片中含有大量的毛蕊花糖苷,扩宽了毛蕊花糖苷的植物来源,白花泡桐属于速生植物,一年甚至可以长三四米高,叶片的生物量大,经济性较高,具有重大的经济价值。
[0070]
为了进一步说明本发明,下面通过以下实施例进行详细说明。本发明以下实施例中所用的实验原料的来源没有特殊的限制,从市场上购买或者按照本领域技术人员熟知的常规制备方法制备得到的即可。
[0071]
下述实施例中高效液相检测条件如下:
[0072]
色谱柱:agilent zorbax hc-c18(2)(4.6mm
×
250mm,5μm);
[0073]
柱温:30℃;
[0074]
进样量:10μl;
[0075]
流速:1ml/min;
[0076]
检测波长:254nm;
[0077]
流动相:甲醇(b)-0.1vol%甲酸缓冲溶液(a),梯度洗脱,见下表1:
[0078]
表1
[0079]
时间(min)a(vol%)b(vol%)0901086832445050520100650100669010769010
[0080]
实施例1
[0081]
称取白花泡桐叶粉末0.5g,置于具塞锥形瓶中,精密加入70vol%甲醇10ml,密塞,称定质量,超声处理(功率500w,频率40khz)30min,放冷,用70vol%甲醇补足减失的质量,取上清液用0.45μm微孔滤膜滤过,制得白花泡桐叶样品溶液。
[0082]
对白花泡桐叶样品溶液进行hplc检测并记录其指纹图谱,色谱条件为:色谱柱:1号柱子:agilent zorbax hc-c18(2)(4.6mm
×
250mm,5μm);2号柱子:waters xbridge c18
(4.6mm
×
150mm,3.5μm);3号柱子:wondacract c-18wrs(4.6mm
×
250mm,5μm)。;流动相:甲醇(b)-0.1vol%甲酸缓冲溶液(a);检测波长:254nm;流速:1ml/min;柱温:30℃;进样量:10μl;梯度洗脱,具体洗脱程序如下表所示:
[0083]
梯度洗脱程序
[0084]
时间(min)a(vol%)b(vol%)0901086832445050520100650100669010769010
[0085]
结果如图5所示,采用1号色谱柱分离,各峰分离度较好,色谱峰多且色谱峰形相对较好。因此,选定agilent zorbax hc-c18(2)(4.6mm
×
250mm,5μm)作为色谱柱。
[0086]
实施例2
[0087]
参考实施例1的分离方法,考察了甲醇-水系统、甲醇-0.1vol%甲酸系统、甲醇-0.1vol%乙酸系统、甲醇-0.1vol%磷酸系统作为流动相对于白花泡桐叶hplc色谱图的影响。
[0088]
结果如图6所示,采用甲醇-水系统、甲醇-0.1vol%乙酸系统、甲醇-0.1vol%磷酸系统,很多色谱峰都无法分离且重叠色谱峰多,而甲醇-0.1vol%甲酸系统的分离效果是最好的,色谱峰形好,色谱出峰的时间合适。因此选择甲醇-0.1vol%甲酸系统作为流动相。
[0089]
实施例3
[0090]
参考实施例1的分离方法,考察了检测波长为210nm、254nm、270nm、290nm、320nm和365nm时得到白花泡桐叶hplc色谱图。
[0091]
结果如图7所示,在254nm波长下的各个色谱峰均具有较好的紫外光吸收,其色谱信息较丰富,基线较平稳,因此选择254nm作为白花泡桐叶的测定波长。
[0092]
实施例4
[0093]
参考实施例1的分离方法,考察了色谱柱温度(25℃,30℃和35℃)对白花泡桐叶hplc色谱图的影响。
[0094]
结果如图8所示,在其它的色谱条件一致的情况下,色谱柱温度为30℃时,各峰的分离效果最佳,因此,确定柱温为30℃。
[0095]
实施例1中,采用1号色谱柱分离的白花泡桐叶样品的hplc指纹图谱如图2所示,其中图谱呈现23个特征峰,其中11号峰为共有峰,强度较高,保留时间相对居中与其余色谱峰的分离度良好,因此选定11号峰为参照峰。因不确定该峰所代表的化合物,对其进行快速制备及鉴定。鉴定方法在前面已经详细说明,在此便不再一一赘述。经鉴定,11号峰为毛蕊花糖苷,鉴于此,本发明提供一种从泡桐叶中提取毛蕊花糖苷的方法,并针对相应参数进行了一系列优化,如下所述。
[0096]
实施例5
[0097]
本实施例用于考察不同的提取溶剂对从泡桐叶中提取的毛蕊花糖苷含量的影响:
[0098]
取白花泡桐叶0.5g,以20倍量70vol%乙醇、50vol%乙醇、30vol%乙醇、水、50vol%甲醇和30vol%甲醇分别在70℃下超声提取2h,取滤液,浓缩干燥,得到粗提物。
[0099]
经测定,结果表明70vol%乙醇、50vol%乙醇、30vol%乙醇、水、50vol%甲醇、30vol%甲醇提取毛蕊花糖苷的含量分别为5.34%、10.59%、9.83%、4.79%、10.21%、8.24%,由此可知在30~50vol%的乙醇溶液的提取率均较高,50vol%乙醇的提取率最高。
[0100]
实施例6
[0101]
本实施例用于考察提取方式对从泡桐叶中提取的毛蕊花糖苷含量的影响:
[0102]
回流提取:取白花泡桐叶0.5g,以20倍量50%乙醇70℃回流提取2h,滤液浓缩干燥,得到粗提物;
[0103]
超声提取:取白花泡桐叶0.5g,以20倍量50%乙醇70℃超声提取2h,滤液浓缩干燥,得到粗提物。
[0104]
测定粗提物中毛蕊花糖苷的含量,结果表明超声提取与回流提取的毛蕊花糖苷的含量分别为11.18%和8.26%,提取量都较高,但在提取一次的情况下超声提取的毛蕊花糖苷含量更多。
[0105]
实施例7
[0106]
本实施例用于考察提取料液比对从泡桐叶中提取的毛蕊花糖苷含量的影响:
[0107]
取白花泡桐叶0.5g,以料液比为(白花泡桐叶:50vol%乙醇的体积,g/ml)1:5、1:10、1:20、1:30和1:50,加入50vol%乙醇70℃回流提取2h,滤液浓缩干燥,得到粗提物。
[0108]
测定不同提取料液比下提取的毛蕊花糖苷的含量。结果表明,料液比1:5g/ml、1:10g/ml、1:20g/ml、1:30g/ml、1:50g/ml提取毛蕊花糖苷的含量分别为5.85%、7.2%、9.89%、10.45%、10.46%,由此可知在料液比为1:(20~50)g/ml时,提取的毛蕊花糖苷含量均较高。但在高于1:30g/ml之后的提取量增加不多,因此从节省溶剂的角度考虑,料液比为1:30g/ml是更优选择。
[0109]
实施例8
[0110]
本实施例用于考察50vol%乙醇的提取时间对从泡桐叶中提取的毛蕊花糖苷含量的影响:
[0111]
取白花泡桐叶0.5g,以20倍量50vol%乙醇在70℃下超声提取30min、60min、90min、120min后,滤液浓缩干燥,得到粗提物。
[0112]
测定不同提取时间下,毛蕊花糖苷的含量。结果表明,提取30min、60min、90min、120min毛蕊花糖苷的含量分别为9.99%、10.97%、11.06%、11.25%,由此可知在0.5~2h的提取得率均较高,且随着提取时间的延长,提取得率逐渐增加,但在1h后提取得率并没有大幅度增加,为节约成本最优选可采用1h。
[0113]
实施例9
[0114]
本实施例用于考察水提取时间对从泡桐叶中提取的毛蕊花糖苷含量的影响:
[0115]
取白花泡桐叶0.5g,以30倍量水在70℃下超声提取30min、60min、90min、120min、150min,滤液浓缩干燥,得到粗提物。
[0116]
测定不同提取时间的毛蕊花糖苷的含量,结果表明提取30min、60min、90min、120min、150min毛蕊花糖苷的含量分别为7.25%、7.71%、8.13%、8.22%、8.49%,由此可知在30~150min的提取得率均较高,且随着提取时间的延长,提取得率逐渐增加,但在
90min后提取得率并没有大幅度增加,为节约成本最优选可采用90min。
[0117]
实施例10
[0118]
本实施例用于考察水提取次数对从泡桐叶中提取的毛蕊花糖苷含量的影响:
[0119]
取白花泡桐叶0.5g,以30倍量水70℃超声提取1次、2次、3次,每次提取2h,滤液浓缩干燥,得到粗提物。
[0120]
测定不同提取次数的毛蕊花糖苷的含量,结果表明提取1次、2次、3次时毛蕊花糖苷的含量分别为7.25%、10.35%、10.54%。由此随提取次数的增加,提取的毛蕊花糖苷的含量也逐渐增加,但在提取次数为3次的情况下,提取含量增加不明显,因此可选择提取两次来适当增加采用水作为提取溶剂时提取得到的毛蕊花糖苷的含量。
[0121]
实施例11
[0122]
本实施例考察大孔吸附树脂的种类对得到的毛蕊花糖苷富集部位中毛蕊花糖苷含量的影响:
[0123]
在装有2.0g已处理好的ab-8、dm-301、da-201、d-101、hpd100、dm-130大孔吸附树脂的锥形瓶中分别加入100ml白花泡桐叶粗提物溶液。25℃振荡吸附6h,每隔0.5h取上清液1ml,采用hplc测定,计算吸附率[吸附率=(吸附前的质量浓度-吸附后的质量浓度)/吸附前的质量浓度]。取已吸附好的树脂,经适量水洗后加入50vol%乙醇50ml,恒温振荡解吸附,取上清液1ml,加10vol%甲醇定容至10ml。采用hplc测定,计算解吸率[解吸率=解吸附后质量浓度/(吸附前的质量浓度-吸附后的质量浓度)]。
[0124]
结果如表2所示,上述几种大孔吸附树脂均能够达到较好的分离纯化效果,但相比而言,d-101与ab-8型大孔吸附树脂对毛蕊花糖苷具有更好的吸附和解吸附效果。另外,d-101大孔吸附树脂相较于ab-8型大孔吸附树脂,具有价格更加低廉的优势。
[0125]
表2
[0126]
树脂型号吸附率(%)解吸率(%)d-10173.2164.54ab-872.7263.89dm-30162.9751.35da-20161.5651.21hpd10060.8851.02dm-13061.3451.22
[0127]
实施例12
[0128]
本实施例考察大孔吸附树脂对粗提物进行分离纯化的过程中,上样液质量浓度对得到的毛蕊花糖苷富集部位中毛蕊花糖苷含量的影响:
[0129]
精密称取3份处理好的大孔吸附树脂(每份40g)分别湿法装柱。将上样液质量浓度分别为15mg/ml、25mg/ml、35mg/ml的样品溶液(经母液稀释可得)以相同的体积流量(2bv/h)过大孔树脂柱。吸附完全后,采用hplc测定,计算吸附率[吸附率=(吸附前的质量浓度-吸附后的质量浓度)/吸附前的质量浓度]。
[0130]
结果表明,在上样液质量浓度为15mg/ml、25mg/ml、35mg/ml下,样品溶液的吸附率分别为72.44%、73.32%、72.56%。当质量浓度为25mg/ml时,吸附率达到最大值。
[0131]
实施例13
[0132]
本实施例考察大孔吸附树脂对粗提物进行分离纯化的过程中,洗脱剂对得到的毛蕊花糖苷富集部位中毛蕊花糖苷含量的影响:
[0133]
精密称取4份处理好的大孔吸附树脂(每份40g)分别湿法装柱上样(上样液为25mg/ml的粗提物溶液),用3bv的蒸馏水洗脱除杂后,分别用3bv体积分数为10%、30%、50%、70%的乙醇溶液以2bv/h的体积流量进行洗脱,采用hplc测定,计算解吸率[解吸率=解吸后质量浓度/(吸附前的质量浓度-吸附后的质量浓度)]。
[0134]
结果表明当用体积分数为10%、30%、50%、70%的乙醇溶液进行洗脱,洗脱的解吸率分别为17.25%、41.63%、98.16%、98.58%。结果表明体积分数为10%、30%的乙醇洗脱不完全,体积分数为50%、70%的乙醇可将毛蕊花糖苷几乎完全洗脱下来,但70vol%乙醇将毛蕊花糖苷洗脱下来的同时,杂质增多,成本增大。综合考虑,50vol%的乙醇作为洗脱剂是更优选择。
[0135]
实施例14
[0136]
本实施例考察大孔吸附树脂对粗提物进行分离纯化的过程中,洗脱剂用量对得到的毛蕊花糖苷富集部位中毛蕊花糖苷含量的影响:
[0137]
称取已处理好的大孔吸附树脂40g湿法装柱上样(上样液为25mg/ml的粗提物溶液),用3bv蒸馏水洗脱除杂后,用50vol%乙醇洗脱,流出液每1bv(20ml)收集1份。采用hplc测定,计算洗脱率(洗脱率=洗脱下来的质量/洗脱前的质量)。
[0138]
洗脱剂体积为1bv、2bv、3bv、4bv、5bv、6bv时的洗脱率分别为29.49%、43.49%、21.36%、4.45%、0.42%和0.11%。结果表明,洗脱至3~4.0bv时,即可将98%以上的毛蕊花糖苷洗脱下来,综合考虑成本,50vol%乙醇的洗脱用量优选为3~4.0bv,更优选为4.0bv。
[0139]
实施例15
[0140]
本实施例考察大孔吸附树脂对粗提物进行分离纯化的过程中,洗脱剂的体积流量对得到的毛蕊花糖苷富集部位中毛蕊花糖苷含量的影响:
[0141]
称取已处理好的d-101型大孔吸附树脂40g湿法装柱上样(上样液为25mg/ml的粗提物溶液),先以3bv蒸馏水洗脱除杂,再分别用3bv的50vol%乙醇以1bv/h、2bv/h、3bv/h、4bv/h、5bv/h、6bv/h体积流量洗脱,流出液每1bv收集1次。采用hplc测定,计算洗脱率(洗脱率=洗脱下来的质量/洗脱前的质量)。
[0142]
洗脱剂体积为1bv/h、2bv/h、3bv/h、4bv/h、5bv/h、6bv/h时的洗脱率分别为97.78%、95.32%、94.69%、91.53%、89.77%、87.41%。结果表明,洗脱体积流量为1bv/h、2bv/h时有较高的洗脱率。若综合考虑洗脱效果与时间成本,则洗脱剂的最优体积流量为2bv/h。
[0143]
实施例16
[0144]
本实施例采用水提取泡桐叶中的毛蕊花糖苷,具体步骤如下:
[0145]
将0.82kg白花泡桐干叶放在30l的不锈钢桶中,加入30l纯净水溶液,70℃条件下进行超声提取,提取时间为2h。提取结束后,将提取液滤出,再次加入30l纯净水溶液进行超声提取,合并滤液,浓缩干燥,得到白花泡桐叶粗提物74.3g,得率为9.06%。经过高效液相检测,水提取的白花泡桐叶粗提物中,毛蕊花糖苷的纯度为10.052%。
[0146]
实施例17
[0147]
本实施例采用50vol%的乙醇水溶液提取泡桐叶中的毛蕊花糖苷,具体步骤如下:
[0148]
将0.94kg白花泡桐干叶放在30l的不锈钢桶中,加入20l浓度为50vol%的乙醇水溶液,超声提取,提取时间为1h。提取结束后,将提取液滤出,得到白花泡桐叶粗提物104.7g,得率为11.1%。经过高效液相检测,50vol%的乙醇提取的白花泡桐叶粗提物中,毛蕊花糖苷的纯度为10.514%。
[0149]
对比实施例1~2得到的粗提物中,毛蕊花糖苷的纯度的相差不大。后续的提取纯化以实施例1得到的粗提物进行。
[0150]
实施例18
[0151]
本实施例采用树脂对实施例16得到的粗提物进行分离纯化,得到毛蕊花糖苷富集部位,具体步骤如下:
[0152]
取55g实施例1得到的白花泡桐叶粗提物,溶解于2.2l水中,上d101大孔吸附树脂,首先用水溶液进行洗脱,洗脱体积为3个柱体积,流速为2个柱体积/小时,然后用体积分数为10%的乙醇水溶液进行洗脱,洗脱体积为3个柱体积,流速为2个柱体积/小时,再用体积分数为30%的乙醇水溶液进行洗脱,洗脱体积为3个柱体积,流速为2个柱体积/小时,最后用体积分数为50%的乙醇水溶液进行洗脱,洗脱体积为4个柱体积,流速为2个柱体积/小时。得到毛蕊花糖苷富集部位,重量为3.03g,得率为5.51%。经过高效液相检测,毛蕊花糖苷的纯度达到41.6%。
[0153]
本实施例中的大孔吸附树脂采用ab-8,dm-301,da-201,hpd100、dm-130等类型树脂替换后,也可以达到同样效果。
[0154]
实施例19
[0155]
本实施例制备液相色谱(北京京科lc-3000a型制备液相)对得到的毛蕊花糖苷富集部位进一步分离纯化,具体步骤如下:
[0156]
取实施例18得到的毛蕊花糖苷富集部位,进入制备液相色谱仪后得到毛蕊花糖苷收集液,在70℃下进行真空浓缩至固液质量体积比在1:(4~5)g/ml时停止浓缩,然后干燥,得到毛蕊花糖苷固体。经过高效液相检测,以峰面积归一法计算,结果如图9所示,毛蕊花糖苷的纯度达到98.4%。
[0157]
其中,所述制备液相色谱仪的工作条件如下:
[0158]
色谱柱:ymc-pack ods-a(20mm
×
250mm,5μm);
[0159]
流动相:甲醇(b)-水(a)=(33:67,体积比),等度洗脱;
[0160]
检测波长334nm;
[0161]
流动相流速10ml/min;
[0162]
柱温30℃;
[0163]
tr=25.0min;
[0164]
进样量:20μl。
[0165]
所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1