一种3-氧代环丁烷基羧酸的制备方法与流程
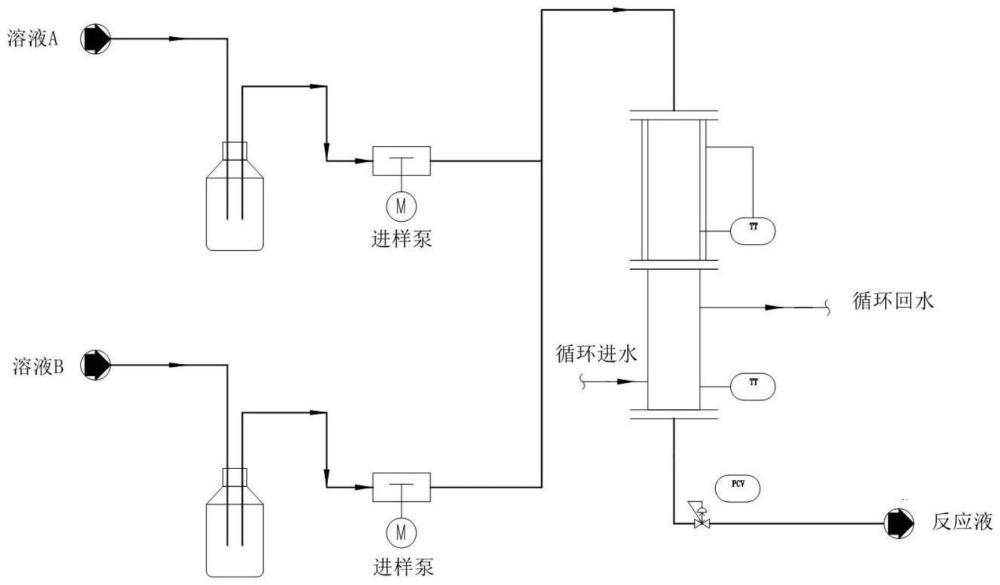
:本发明涉及药物中间体合成领域,具体地说涉及一种3-氧代环丁烷基羧酸的制备方法。
背景技术:
1、3-氧代环丁烷基羧酸是合成很多重要有机中间体的原料,在化学、化工、医药领域有非常广泛的应用。3-氧代环丁烷基羧酸自身也是一个非常重要的医药中间体,被广泛应用于靶向多种自身免疫性疾病、多种抗病毒药物、抗肿瘤药物以及凝血酶抑制剂等靶点的原料药的合成,如ackl抗体,mdm2拮抗剂,jak抑制剂,cetp抑制剂,pde10抑制剂等。同时,3-氧代-1-环丁烷基羧酸衍生得到的一系列化合物也具有比较广泛的应用,例如用来治疗急性髓系白血病的抗癌药物ivosidenib的关键中间体3,3-二氟环丁胺盐酸盐,需要使用3-氧代环丁烷基羧酸作为起始原料生产制得。
2、cn106866405a公开了一种3-氧代环丁烷基羧酸的制备方法,该方法以3,3-二甲氧基环丁烷-1,1-二羧酸二异丙酯为合成原料,在一定浓度的强碱溶液中室温反应4小时,然后加入一定体积的浓盐酸,继续回流反应24~48小时。该方法的产物收率仅能达到80%,收率不甚理想,且反应时间较长,在工业化生产时会大幅增加生产周期。
3、
4、cn113527081a公开了一种3-氧代环丁烷基羧酸的制备方法,该方法以将3,3-二甲氧基环丁烷-1,1-二羧酸二异丙酯与相转移催化剂甲基三辛基氯化铵混合在盐酸中,70~80℃加热回流12小时,随后重结晶得到3-氧代环丁烷基羧酸,产率86%。与cn106866405a中公开的方法相比小幅缩短了反应时间,但产率并未获得较大提高。
5、
6、wo2007062308 a2公开了一种3-氧代环丁烷基羧酸的制备方法,将3,3-二甲氧基环丁烷-1,1-二羧酸二异丙酯与盐酸混合,在155-160℃条件下微波照射1小时。经过萃取、过滤、浓缩后得到3-氧代环丁烷基羧酸,产率可达100%。但该反应为克级反应,规模较小,大规模生产对设备有较高要求。
7、
8、通过查阅相关文献及专利发现,现有技术中3-氧代环丁烷基羧酸的制备方法存在不适用于工业化生产的问题,因此有必要开发并优化能够减少反应时间、提高反应收率、安全稳定的反应路线,并且适合工业放大生产的制备方法。
技术实现思路
1、发明目的:本发明的目的在于提供一种3-氧代环丁烷基羧酸的连续化合成方法,以解决现有的3-氧代环丁烷基羧酸合成过程中对设备要求高,反应周期太长等缺陷。
2、一方面,本发明提供了一种式i化合物的制备方法:
3、
4、式ii化合物在酸1及有机溶剂作用下,泵入连续式反应器中进行反应,后经处理制得式i化合物。
5、优选的,所述酸1选自盐酸,硫酸,甲酸,乙酸,氢溴酸或三氟乙酸。
6、优选的,所述酸1的用量为式ii化合物的2~8倍量体积。
7、优选的,所述有机溶剂选自丙酮,甲乙酮,乙二醇二甲醚或乙腈。
8、优选的,所述有机溶剂的用量为式ii化合物的0.2~4倍量体积。
9、优选的,所述的连续式反应器包括但不限于微通道反应器、板式反应器、管式反应器中的一种或多种。其中,管式反应器包括但不限于列管式反应器、含静态混合器的管式反应器或动态管式反应器。
10、优选的,反应液泵入连续式反应器中反应,反应温度范围为150~300℃;反应时间1~40分钟;压力范围为1~5mpa。
11、优选的,反应结束后进行的后处理步骤包括:脱溶、萃杂、脱溶、脱色、萃取、重结晶。
12、有益效果
13、本发明的目的在于克服现有技术中反应过程对设备要求高以及生产周期太长等问题,提供了一种安全、简便,易于放大且生产周期大幅缩短,收率较高,成本较低的工艺。该工艺以式ii化合物作为原料,选用连续式反应装置,使得工业化生产无需采用高压反应釜等复杂设备,即可高效一步制得式i化合物,且过程可控、操作简单、安全性高,收率可到85~90%。与现有技术相比,大幅缩短了生产周期、降低了生产成本。综上,本发明提供的技术方案适合工业化生产,具有较高的经济效益。
技术特征:
1.一种式i化合物的制备方法,其特征在于:
2.根据权利要求1所述的制备方法,其特征在于:所述酸1选自盐酸,硫酸,甲酸,乙酸,氢溴酸或三氟乙酸。
3.根据权利要求1所述的制备方法,其特征在于:所述酸1的用量为式ii化合物的2~8倍量体积。
4.根据权利要求1所述的制备方法,其特征在于:所述的有机溶剂选自丙酮,甲乙酮,乙二醇二甲醚或乙腈。
5.根据权利要求1所述的制备方法,其特征在于:所述的有机溶剂的用量为式ii化合物的0.2~4倍量体积。
6.根据权利要求1所述的制备方法,其特征在于:所述的连续式反应器选自微通道反应器、板式反应器、管式反应器中的一种或多种。
7.根据权利要求6所述的制备方法,其特征在于:所述的管式反应器选自列管式反应器、含静态混合器的管式反应器或动态管式反应器。
8.根据权利要求1所述的制备方法,其特征在于:反应液泵入连续式反应器中反应,反应温度范围为150~300℃;反应时间1~40分钟;压力范围为1~5mpa。
9.根据权利要求1所述的制备方法,其特征在于:反应结束后进行的后处理步骤包括:脱溶、萃杂、脱溶、脱色、萃取、重结晶。
技术总结
本发明公开了一种3‑氧代环丁烷基羧酸(式I化合物)的制备方法,该方法采用连续式反应装置,以3,3‑二甲氧基环丁烷‑1,1‑二羧酸二异丙酯(式II化合物)作为原料,与盐酸混合,在高温加压条件下一步反应制得反式‑环丁烷‑1,2‑二羧酸。该工艺安全、简便,易于放大且收率较高,成本较低,可实现连续性大规模生产制备。
技术研发人员:马广超,张峰,李伟,李志远,田浩,洪松
受保护的技术使用者:南京药石科技股份有限公司
技术研发日:
技术公布日:2024/5/8
- 还没有人留言评论。精彩留言会获得点赞!