一种丙烯酸酯类衍生物-13C的制备方法与流程
本发明涉及丙烯酸酯类衍生物,具体涉及一种丙烯酸酯类衍生物-13c的制备方法。
背景技术:
1、丙烯酸酯类衍生物是重要的高分子单体和基本有机化工原料。其聚合物化学性质稳定,可以满足不同药物剂型的需要,其在药物制剂方面得到了广泛的应用。由于丙烯酸及其衍生物丙烯酰氯化学性质活泼,在空气、光照条件下易聚合,造成采用传统酯缩合方式合成同位素标记碳元素丙烯酸酯类衍生物的合成产率较低。
2、diemduyle(j.labelled cpd.radiopharm.43,1107-1111(2000))等报道了采用乙烯基溴化镁与14c标记二氧化碳反应得到14c标记丙烯酸,并采用其合成得到14c标记丙烯酸甲酯,产率较低。礼来公司(wo2021/118877)报道了采用乙烯基溴化镁与13c标记二氧化碳反应合成得到13c标记丙烯酸,亦存在产率较低的问题。
3、现有的合成同位素标记丙烯酸方法主要有:
4、1.diemduyle(j.labelled cpd.radiopharm.43,1107-1111(2000))等报道了采用乙烯基溴化镁与14c标记二氧化碳反应得到14c标记丙烯酸,并采用其合成得到14c标记丙烯酸甲酯,按其比活度计算,产率为24%,其产率较低。
5、2.礼来公司(wo2021/118877)报道了采用乙烯基溴化镁与13c标记二氧化碳反应合成得到13c标记丙烯酸,其产率为24%,同样存在产率过低的问题。基于此,本发明提供了一种丙烯酸酯类衍生物-13c的制备方法。
技术实现思路
1、针对现有技术的缺陷,本发明的目的是提供一种丙烯酸酯类衍生物-13c的制备方法,以解决上述背景技术中提出的问题。
2、本发明解决技术问题采用如下技术方案:
3、本发明提供了一种丙烯酸酯类衍生物-13c的制备方法,包括以下步骤:
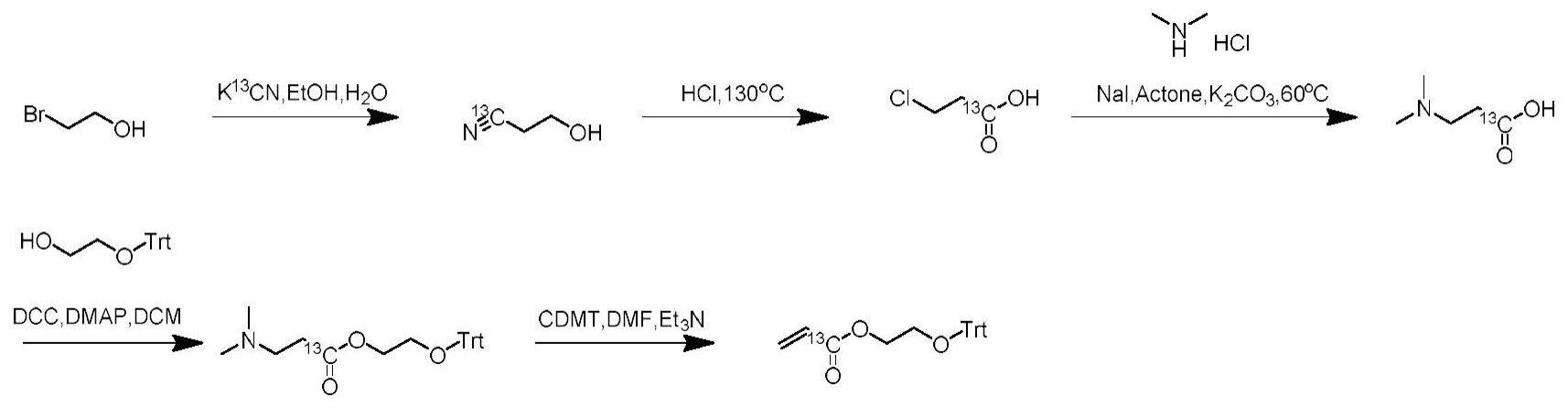
4、步骤一:3-羟基丙腈-1-13c粗品的制备;
5、步骤二:将3-羟基丙腈-1-13c粗品置于闷罐中,制备反应成n,n-二甲基-β-丙氨酸-1-13c粗品;
6、步骤三:以n,n-二甲基-β-丙氨酸-1-13c粗品为底料,合成反应成丙烯酸酯类衍生物-13c。
7、优选地,所述3-羟基丙腈-1-13c粗品的制备中具体的反应过程为:
8、s1:氮气保护下,25ml单口烧瓶中,加入2-溴乙醇、k13cn、去离子水、无水乙醇,升温至80℃搅拌6小时;
9、s2:氰根检测显示原料反应完全,反应液降至室温,浓缩除去溶剂,加入5ml的丙酮,室温搅拌20分钟,过滤,收集滤液,浓缩除去溶剂,得到3-羟基丙腈-1-13c粗品。
10、优选地,所述2-溴乙醇、k13cn、去离子水和无水乙醇的加入量分别为524.8mg、132mg、1.6ml和3.2ml。
11、优选地,所述3-羟基丙腈-1-13c的合成过程中,除可采用2-溴乙醇作为原料,还采用2-氯乙醇作为起始原料。
12、优选地,所述n,n-二甲基-β-丙氨酸-13c粗品的制备反应过程为:
13、s01:将3-羟基丙腈-1-13c粗品置于闷罐中,加入4ml的浓盐酸,升温至130℃反应7小时,加入8ml水,乙酸乙酯萃取,浓缩除去溶剂得到3-氯丙酸-1-13c粗品;
14、s02:将3-氯丙酸-1-13c粗品置于25ml单口烧瓶中,加入二甲胺盐酸盐、碘化钠、碳酸钾、丙酮、室温搅拌20分钟,随后升温至60℃反应6小时,tlc检测原料反应完全,浓缩除去溶剂;
15、s03:加入10ml的水,采用1n盐酸水溶液调节体系ph至6-7,浓缩除水,得到n,n-二甲基-β-丙氨酸-13c粗品。
16、优选地,所述二甲胺盐酸盐、碘化钠、碳酸钾、丙酮的加入量分别为489mg、90mg、1.1g和5ml。
17、优选地,所述丙烯酸酯类衍生物-13c为2-(三苯基甲氧基)乙基丙烯酸酯-1-13c、2-(苄氧基)乙基丙烯酸酯-113c和3-(叔丁氧基)-3-氧代丙基丙烯酸酯-1-13c中的一种。
18、优选地,所述2-(三苯基甲氧基)乙基丙烯酸酯-1-13c的合成反应方法为:
19、s11:将n,n-二甲基-β-丙氨酸-1-13c粗品置于25ml单口烧瓶中,加入2-(三苯甲氧基)乙醇,4-二甲氨基吡啶和二氯甲烷,室温搅拌20分钟,随后加入n,n'-二环己基碳酰亚胺,反应液室温搅拌16小时;
20、s12:tlc显示n,n-二甲基-β-丙氨酸-1-13c反应完全,加入二氯甲烷(20ml),饱和氯化铵水溶液洗涤,有机相无水硫酸钠干燥,柱层析纯化得到2-(三苯基甲氧基)乙基3-(二甲基氨基)丙酸酯-1-13c粗品;
21、s13:将2-(三苯基甲氧基)乙基3-(二甲基氨基)丙酸酯-1-13c粗品置于25ml单口烧瓶中,加入6ml的n,n-二甲基甲酰胺溶解,随后加入125mg的2-氯-4,6-二甲氧基-1,3,5-三嗪,74mg的三乙胺,加毕,室温反应30分钟,tlc显示原料反应完全,加入20ml的水,乙酸乙酯萃取,有机相采用饱和食盐水洗涤,柱层析得到2-(三苯基甲氧基)乙基丙烯酸酯-1-13c。
22、优选地,所述2-(三苯甲氧基)乙醇、4-二甲氨基吡啶、二氯甲烷、n,n'-二环己基碳酰亚胺的加入量分别为731mg、49mg、8ml和495mg。
23、优选地,所述2-(三苯基甲氧基)乙基丙烯酸酯-1-13c、2-(苄氧基)乙基丙烯酸酯-1-13c和3-(叔丁氧基)-3-氧代丙基丙烯酸酯-1-13c的合成过程中,除可采用2-氯-4,6-二甲氧基-1,3,5-三嗪,三乙胺作为消除试剂外,亦可采用碘甲烷,碳酸氢钠进行霍夫曼消除反应。
24、与现有技术相比,本发明具有如下的有益效果:
25、1、本发明采用n,n-二甲基-β-丙氨酸替代丙烯酸进行酯缩合反应,避免了合成丙烯酸过程以及酯缩合过程中的丙烯酸聚合反应,从而提高了反应产率,填补了国内同位素标记丙烯酸酯类衍生物合成的空白;
26、2、避免了使用气密性要求较高的二氧化碳反应装置,直接使用市面上容易得到的标记氰化钾作为起始原料,反应条件温和,容易实现;
27、3、本发明避免了丙烯酸的自聚反应,提升了反应产率。
技术特征:
1.一种丙烯酸酯类衍生物-13c的制备方法,其特征在于,包括以下步骤:
2.根据权利要求1所述的一种丙烯酸酯类衍生物-13c的制备方法,其特征在于,所述3-羟基丙腈-1-13c粗品的制备中具体的反应过程为:
3.根据权利要求1所述的一种丙烯酸酯类衍生物-13c的制备方法,其特征在于,所述2-溴乙醇、k13cn、去离子水和无水乙醇的加入量分别为524.8mg、132mg、1.6ml和3.2ml。
4.根据权利要求2所述的一种丙烯酸酯类衍生物-13c的制备方法,其特征在于,所述3-羟基丙腈-1-13c的合成过程中,除可采用2-溴乙醇作为原料,还采用2-氯乙醇作为起始原料。
5.根据权利要求1所述的一种丙烯酸酯类衍生物-13c的制备方法,其特征在于,所述n,n-二甲基-β-丙氨酸-13c粗品的制备反应过程为:
6.根据权利要求5所述的一种丙烯酸酯类衍生物-13c的制备方法,其特征在于,所述二甲胺盐酸盐、碘化钠、碳酸钾、丙酮的加入量分别为489mg、90mg、1.1g和5ml。
7.根据权利要求1所述的一种丙烯酸酯类衍生物-13c的制备方法,其特征在于,所述丙烯酸酯类衍生物-13c为2-(三苯基甲氧基)乙基丙烯酸酯-1-13c、2-(苄氧基)乙基丙烯酸酯-113c和3-(叔丁氧基)-3-氧代丙基丙烯酸酯-1-13c中的一种。
8.根据权利要求5所述的一种丙烯酸酯类衍生物-13c的制备方法,其特征在于,所述2-(三苯基甲氧基)乙基丙烯酸酯-1-13c的合成反应方法为:
9.根据权利要求8所述的一种丙烯酸酯类衍生物-13c的制备方法,其特征在于,所述2-(三苯甲氧基)乙醇、4-二甲氨基吡啶、二氯甲烷、n,n'-二环己基碳酰亚胺的加入量分别为731mg、49mg、8ml和495mg。
10.根据权利要求8所述的一种丙烯酸酯类衍生物-13c的制备方法,其特征在于,所述2-(三苯基甲氧基)乙基丙烯酸酯-1-13c、2-(苄氧基)乙基丙烯酸酯-1-13c和3-(叔丁氧基)-3-氧代丙基丙烯酸酯-1-13c的合成过程中,除可采用2-氯-4,6-二甲氧基-1,3,5-三嗪,三乙胺作为消除试剂外,亦采用碘甲烷,碳酸氢钠进行霍夫曼消除反应。
技术总结
本发明公开了一种丙烯酸酯类衍生物‑<supgt;13</supgt;C的制备方法,包括以下步骤:步骤一:3‑羟基丙腈‑1‑<supgt;13</supgt;C粗品的制备;步骤二:将3‑羟基丙腈‑1‑<supgt;13</supgt;C粗品置于闷罐中,制备反应成N,N‑二甲基‑β‑丙氨酸‑1‑<supgt;13</supgt;C粗品;步骤三:以N,N‑二甲基‑β‑丙氨酸‑1‑<supgt;13</supgt;C粗品为底料,合成反应成丙烯酸酯类衍生物‑<supgt;13</supgt;C。本发明采用N,N‑二甲基‑β‑丙氨酸替代丙烯酸进行酯缩合反应,避免了合成丙烯酸过程以及酯缩合过程中的丙烯酸聚合反应,从而提高了反应产率,填补了国内同位素标记丙烯酸酯类衍生物合成的空白。
技术研发人员:陈晓晖,伍君,谭雅彬,李刚
受保护的技术使用者:长沙贝塔医药科技有限公司
技术研发日:
技术公布日:2024/1/12
- 还没有人留言评论。精彩留言会获得点赞!