一种同时制备三丙基乙腈、三丙基酰胺、三丙基乙酸的方法与流程
本发明涉及三丙基乙腈制备,尤其涉及一种同时制备三丙基乙腈、三丙基酰胺、三丙基乙酸的方法。
背景技术:
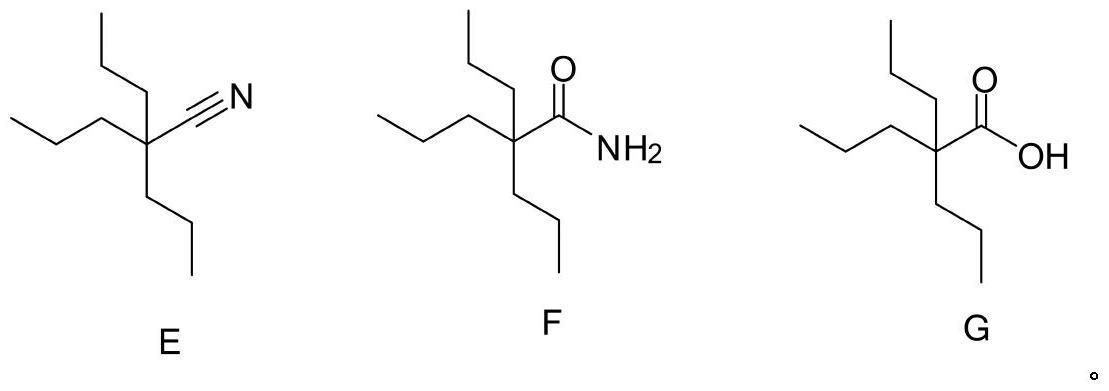
1、三丙基乙腈(化合物e)、三丙基酰胺(化合物f)、三丙基乙酸(化合物g)是丙戊酸钠原料药合成工艺中需要研究的工艺杂质,同时欧洲药典9.0中明确指出需要对这三中杂质进行研究,对于丙戊酸钠原料药生产中有关物质的检测和控制起到关键的作用,结构式如下:
2、
3、在中间体2-氰基-2-丙基戊酸甲酯(化合物b)的制备中,专利us4127604以氰乙酸乙酯、溴丙烷、丙醇钠为原料制备化合物化合物h,同时丙醇钠是用金属钠和丙醇反应制备得到的,试剂不常见,由于采用丙醇钠催化,往往会发生酯交换反应,生成副产物i,不利于产物的纯化和产物质量的中控。
4、
5、在关键中间体丙戊腈(化合物d)的制备过程中,专利us4127604需要经过两次蒸馏才得到合格化合物d,能耗较大。
6、在制备化合物e中,现有技术“process for introducing alkyl radicals intocarbon chains having a functional group and compounds prepared by saidprocess;us4377533”根据专利中的example 22制备三丙基戊腈主要是通过乙腈和氨基钠、异丁醇钠进行碳负离子化,然后与卤代烷烃进行烷基化反应生成三丙基戊腈。
7、根据专利us 4377533中example 1、example 18、example 23的合成工艺分析,本工艺缺陷如下:(1)烷基化工艺使用到氨基钠,试剂的危险性相对较大;(2)乙腈进行烷基化反应容易生产单取代产物戊腈,二取代产物丙戊腈,因此副反应较多,副产物与主产物性质相近,分离困难;
8、
9、在氰基水解成羧酸的过程中,大多数传统工艺采用硫酸和亚硝酸钠试剂进行催化,如专利us4127604,反应中过量的亚硝酸钠转化为亚硝酸,进一步分解成有毒气体一氧化氮和二氧化氮污染大气、腐蚀设备。
技术实现思路
1、本发明的目的是针对现有技术中的不足,提供一种同时制备三丙基乙腈、三丙基酰胺、三丙基乙酸的方法。
2、为实现上述目的,本发明采取的技术方案是:
3、提供一种同时制备三丙基乙腈、三丙基酰胺、三丙基乙酸的方法,包括如下步骤:
4、步骤一,在反应器中加入氰乙酸甲酯(化合物a)、甲醇和溴丙烷,控制反应温度50~65℃,滴加甲醇钠的甲醇溶液,反应结束,后处理,得到2-氰基-2-丙基戊酸甲酯;
5、步骤二,在氢氧化钠水溶液中加入步骤一制备得到的2-氰基-2-丙基戊酸甲酯(化合物b),升温至50~70℃反应,反应结束,降温至室温,调节ph,分相,得到油相为2-氰基-2-丙基戊酸粗品(化合物c);
6、步骤三,将步骤二得到的2-氰基-2-丙基戊酸粗品加入反应瓶中,安装常压蒸馏装置,收集气相温度为160~180℃的馏分,得到丙戊腈(化合物d);
7、步骤四,在反应器中加入碱性试剂、thf、丙戊腈和碘丙烷,氮气置换多次,升温至50~70℃,反应结束后进行后处理,得到三丙基乙腈(化合物f);
8、步骤五,在反应器中加入硫酸水溶液,加入三丙基乙腈(化合物e),搅拌,升温至130~150℃,反应20-35小时,反应结束降温至室温,分离、纯化,分别得到三丙基乙酰胺(化合物f)和三丙基乙酸(化合物g)。
9、
10、进一步地,所述步骤一中,氰乙酸甲酯与溴丙烷的摩尔比为1:(2~3),氰乙酸甲酯与甲醇钠的摩尔比为1:(2~3)。
11、进一步地,所述步骤一中,后处理具体步骤为:反应结束后常压蒸馏甲醇,得到固液混合物,加入乙酸异丙酯,过滤除盐,盐用乙酸异丙酯漂洗、洗液和滤液合并,水洗,油相浓缩、蒸馏。
12、进一步地,所述步骤二中,氢氧化钠与2-氰基-2-丙基戊酸甲酯的摩尔比为1:(1~4.5);反应时间为5-8小时;加入盐酸调节ph为1-1.5。
13、进一步地,所述步骤三中,安装常压蒸馏装置后,缓慢升温至140℃,之后逐步升温至205℃,收集气相温度为160~180℃的馏分。
14、进一步地,所述步骤四中,碱性试剂为氢化钠、氢化钙、氢化锂铝、丁基锂中的一种或几种;碱性试剂与丙戊腈的摩尔比为1:(0.3~1);反应时间为7-15小时。
15、进一步地,所述步骤四中,后处理具体步骤为:反应液缓慢加入水焠灭,控制温度50℃以下,移出上清液,上清液减压浓缩,加入甲苯,油相使用氢氧化钠水溶液萃取、水洗、浓盐酸洗、水洗,分相;油相使用水泵减压至0.95mpa旋蒸,得到的油相倒入单口瓶中,使用水泵减压至0.095mpa蒸馏,收集气相温度为138℃的馏分,得到馏分三丙基乙腈。
16、进一步地,所述步骤五中,硫酸水溶液的质量浓度为50-70%。
17、进一步地,所述步骤五中,分离、纯化的具体步骤为:加入甲苯和水稀释,过滤,分相,油相水洗,油相加入氢氧化钠水溶液升温至回流,回流20-30分钟,降温至室温,用二氯甲烷萃取,分相,得到三丙基乙酰胺的二氯甲烷溶液,以及三丙基乙酸钠水相;三丙基乙酸钠水相加入浓盐酸酸化为ph=1,用二氯甲烷反萃,分相,二氯甲烷相水浴30℃减压浓缩干,使用油泵60℃减压抽1小时,得到三丙基乙酸;三丙基乙酰胺的二氯甲烷相使用旋转蒸发仪30~70℃减压浓缩至干,使用油泵在70℃下减压抽0.5小时,得到三丙基乙酰胺。
18、本发明采用以上技术方案,与现有技术相比,具有如下技术效果:
19、本发明在制备化合物b的时候采用甲醇钠和甲醇体系,原料易得,因此也不会出现专利us4127604中的酯交换副产物i的生成,减少了副反应;同时避免了使用丙醇和金属钠的反应制备丙醇钠,降低了实验操作的危险性。
20、本发明在制备化合物d时候,直接把蒸馏装置安装在反应瓶上,做到了边反应边蒸馏的效果,避免了专利us4127604中的二次蒸馏,降低了能耗。
21、本发明在制备化合物e过程中,使用丙戊腈和溴丙烷进行烷基化反应,使用氢化钠催化,相对于专利us 4377533中使用的使用乙腈和溴丙烷进行烷基化,氨基钠碱试剂催化而言,试剂的危险性大大降低,同时减少了副产物戊腈和丙戊腈的生成,副反应也明显减少。
22、本发明使用硫酸水溶液在130~150℃催化,将氰基水解为酰胺和羧酸的混合物,避免了有毒有害气体一氧化氮和二氧化氮气体的生成;一条合成路线同时制备丙戊酸钠三个高纯度有机杂质,即化合物e、f、g纯度在98%以上,大大提高了杂质的制备效率。
技术特征:
1.一种同时制备三丙基乙腈、三丙基酰胺、三丙基乙酸的方法,其特征在于,包括如下步骤:
2.根据权利要求1所述的同时制备三丙基乙腈、三丙基酰胺、三丙基乙酸的方法,其特征在于,所述步骤一中,氰乙酸甲酯与溴丙烷的摩尔比为1:(2~3),氰乙酸甲酯与甲醇钠的摩尔比为1:(2~3)。
3.根据权利要求1所述的同时制备三丙基乙腈、三丙基酰胺、三丙基乙酸的方法,其特征在于,所述步骤一中,后处理具体步骤为:反应结束后常压蒸馏甲醇,得到固液混合物,加入乙酸异丙酯,过滤除盐,盐用乙酸异丙酯漂洗、洗液和滤液合并,水洗,油相浓缩、蒸馏。
4.根据权利要求1所述的同时制备三丙基乙腈、三丙基酰胺、三丙基乙酸的方法,其特征在于,所述步骤二中,氢氧化钠与2-氰基-2-丙基戊酸甲酯的摩尔比为1:(1~4.5);反应时间为5-8小时;加入盐酸调节ph为1-1.5。
5.根据权利要求1所述的同时制备三丙基乙腈、三丙基酰胺、三丙基乙酸的方法,其特征在于,所述步骤三中,安装常压蒸馏装置后,缓慢升温至140℃,之后逐步升温至205℃,收集气相温度为160~180℃的馏分。
6.根据权利要求1所述的同时制备三丙基乙腈、三丙基酰胺、三丙基乙酸的方法,其特征在于,所述步骤四中,碱性试剂为氢化钠、氢化钙、氢化锂铝、丁基锂中的一种或几种;碱性试剂与丙戊腈的摩尔比为1:(0.3~1);反应时间为7-15小时。
7.根据权利要求1所述的同时制备三丙基乙腈、三丙基酰胺、三丙基乙酸的方法,其特征在于,所述步骤四中,后处理具体步骤为:反应液缓慢加入水焠灭,控制温度50℃以下,移出上清液,上清液减压浓缩,加入甲苯,油相使用氢氧化钠水溶液萃取、水洗、浓盐酸洗、水洗,分相;油相使用水泵减压至0.95mpa旋蒸,得到的油相倒入单口瓶中,使用水泵减压至0.095mpa蒸馏,收集气相温度为138℃的馏分,得到馏分三丙基乙腈。
8.根据权利要求1所述的同时制备三丙基乙腈、三丙基酰胺、三丙基乙酸的方法,其特征在于,所述步骤五中,硫酸水溶液的质量浓度为50-70%。
9.根据权利要求1所述的同时制备三丙基乙腈、三丙基酰胺、三丙基乙酸的方法,其特征在于,所述步骤五中,分离、纯化的具体步骤为:加入甲苯和水稀释,过滤,分相,油相水洗,油相加入氢氧化钠水溶液升温至回流,回流20-30分钟,降温至室温,用二氯甲烷萃取,分相,得到三丙基乙酰胺的二氯甲烷溶液,以及三丙基乙酸钠水相;三丙基乙酸钠水相加入浓盐酸酸化为ph=1,用二氯甲烷反萃,分相,二氯甲烷相水浴30℃减压浓缩干,使用油泵60℃减压抽1小时,得到三丙基乙酸;三丙基乙酰胺的二氯甲烷相使用旋转蒸发仪30~70℃减压浓缩至干,使用油泵在70℃下减压抽0.5小时,得到三丙基乙酰胺。
技术总结
本发明公开了一种同时制备三丙基乙腈、三丙基酰胺、三丙基乙酸的方法,以氰乙酸甲酯为起始物料制备三丙基乙腈,解决了反应原料不易得、能耗高、试剂危险、副反应较多、有毒有害气体产生的问题,同时三丙基乙腈进一步酸催化水解,即可得到高纯度的三丙基乙酰胺和三丙基乙酸。
技术研发人员:韦建国,魏正华,顾玉仅,杨益超
受保护的技术使用者:上海青平药业有限公司
技术研发日:
技术公布日:2024/1/12
- 还没有人留言评论。精彩留言会获得点赞!