用于治疗癌症的大环化合物的制作方法
ras是最为人所知的原癌基因之一。大约30%的人类癌症包含三个最显著的成员(kras、hras和nras)的突变,这使它们成为最常见的致癌驱动因素。kras突变通常与不良预后相关,尤其在结直肠癌、胰腺癌、肺癌中。作为最频繁发生突变的ras亚型,kras在过去几年中得到了广泛的研究。在最常见的kras等位基因(包括g12d、g12v、g12c、g13d、g12r、g12a、g12s、q61h等)中,g12c、g12d、g12v占所有k-ras驱动的癌症(包括结直肠癌(crc),胰腺导管腺癌(pdac),肺腺癌(luad))的一半以上。值得注意的是,在所有kras改变的癌症(卵巢的、食管和胃的、子宫的)中,在约7%中也发现了kras野生型扩增,位列改变前列。所有ras蛋白均属于将gtp水解为gdp的小gtp酶的蛋白家族。kras在结构上分为效应子结合叶,然后是变构叶和负责膜锚定的羧基末端区域。效应子叶包括p环、开关i和开关ii区域。开关i/ii环通过介导蛋白-蛋白与效应子蛋白的相互作用,在kras下游信号传导中发挥关键作用,这些效应子蛋白包括丝裂原激活蛋白激酶(mapk)途径中的raf或磷脂酰肌醇3-激酶(p13k)/蛋白激酶b(akt)途径中的pi3k。kras蛋白分别经由结合到gtp和gdp在非活性形式和活性形式之间切换。在生理条件下,这两种状态之间的转变受到鸟嘌呤核苷酸交换因子(gef)的调节,诸如七少之子同源物1(sos1)或涉及催化gdp交换为gtp的gtp酶激活蛋白(gap),从而增强内在gtp酶活性或加速ras介导的gtp水解。响应于细胞外刺激,无活性ras-gdp经转化为活性ras-gtp,活性ras-gtp直接与raf ras结合域(rafrbd)结合,从而将raf激酶家族从细胞质招募到细胞膜,在细胞膜中使它们二聚化并且变得有活性。活性化的raf随后对其下游丝裂原激活蛋白激酶(mek)和细胞外信号调节激酶(erk)进行一系列磷酸化反应,并传播生长信号。在raf蛋白激酶家族(三种已知亚型araf、braf、craf/raf1)中,braf突变最频繁,并且仍然是mek最有效的活化剂。尽管各个ras和raf家族成员表现出不同的结合偏好,但所有raf都具有用于mapk信号传导的向前传输的保守rbd,频繁地用于表征kras抑制作用(例如本文中的kras-brafrbd)。对于kras,位置12、13、61和146处的突变通过削弱核苷酸水解或使核苷酸交换活性化,导致向活性kras形式转变,从而导致引起肿瘤发生的mapk通路过度活性化。尽管其在癌症恶性肿瘤中的重要性已得到广泛认可,但过去的持续努力未能开发出针对kras突变型癌症的批准的疗法,直到最近,首个选择性药物amg510已快速获批作为kras g12c驱动的非小细胞肺癌(nsclc)的二线治疗。然而,在治疗约6个月后,随着疾病的进展,kras g12c抑制剂的临床获得性耐药性剧烈地出现。所有突变汇聚在一起使ras-mapk信号传导重新活化,其中已经在致癌热点(例如g12/g13/q61)处和开关ii口袋(例如h95、r68和y96)内观察到二次ras突变体;此外,在所有kras突变型或野生型扩增驱动的癌症中,超过85%的癌症仍然缺乏新型药剂。总而言之,无数的逃逸机制和各种致癌等位基因两者都凸显了对额外kras疗法的迫切医疗需求。因此,我们发明了靶向并抑制kras等位基因以用于治疗kras突变型驱动的癌症的口服化合物。
背景技术:
技术实现思路
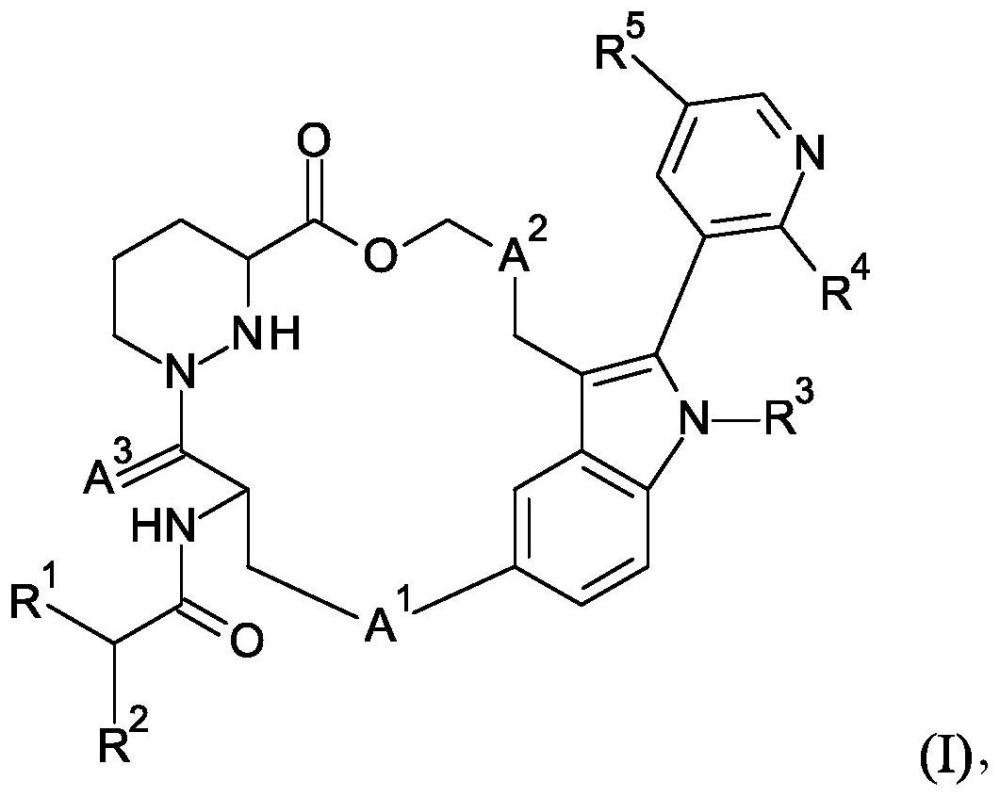
1、本发明涉及具有式(i)的新型化合物,
2、
3、其中
4、r1为或经(二卤代c1-6烷基)羰基取代的6-氧代-2,7-二氮杂螺[4.5]癸烷-7-基;
5、其中r6为经c2-6炔基取代的氮杂环丁烷基,
6、经甲酰基、c2-6炔基、吡啶基c2-6炔基或[(c1-6烷基)2(氧代)-λ6-亚硫基]c1-6烷基羰基取代的环烷基,
7、经独立地选自卤素、(二卤代c1-6烷基)羰基和c2-6炔基的取代基取代一次或两次的哌啶基,或者
8、经(c1-6烷基羰基)羰基、(二卤代c1-6烷基)羰基、c2-6炔基、氰基c1-6烷基、环烷基羰基或三唑基c2-6烯基羰基取代的吡咯烷基;
9、r7为c1-6烷基;
10、r2为c1-6烷基;
11、r3为c1-6烷基或卤代c1-6烷基;
12、r4为c1-6烷氧基c1-6烷基;
13、r5为h、吗啉基、(卤代c1-6烷基)哌嗪基或c1-6烷基哌嗪基;
14、a1为亚噻唑基或亚苯基,所述亚苯基经羟基取代;
15、a2为c1-6亚烷基;
16、a3为o;
17、或其药用盐。
18、本发明还涉及它们的制造、基于根据本发明的化合物的药物及其生产以及其式(i)或(ia)化合物作为kras抑制剂的用途。
19、式(i)或(ia)化合物对g12c、g12d和g12v显示出良好的kras抑制作用。在另一个实施例中,本发明的化合物显示出优异的癌细胞抑制作用。另外,式(i)或(ia)化合物还显示出良好或改善的细胞毒性、溶解度特征。此外,与参考化合物相比,本发明的化合物解决了gsh毒性问题(参见实例27)。
技术特征:
1.一种式(i)化合物,
2.一种式(ia)化合物,
3.根据权利要求1或2所述的化合物,其中r1为经(二卤代c1-6烷基)羰基取代的1-氧代-2,7-二氮杂螺[4.4]壬烷-2-基或经(二卤代c1-6烷基)羰基取代的6-氧代-2,7-二氮杂螺[4.5]癸烷-7-基;其中r6为经(二卤代c1-6烷基)羰基取代的吡咯烷基;r7为c1-6烷基。
4.根据权利要求1至3中任一项所述的化合物,其中r1为经氯(氟)乙酰基取代的1-氧代-2,7-二氮杂螺[4.4]壬烷-2-基或经氯(氟)乙酰基取代的6-氧代-2,7-二氮杂螺[4.5]癸烷-7-基;其中r6为经氯(氟)乙酰基取代的吡咯烷基;r7为甲基。
5.根据权利要求1至4中任一项所述的化合物,其中r1为
6.根据权利要求1至5中任一项所述的化合物,其中r2为异丙基。
7.根据权利要求1至6中任一项所述的化合物,其中r3为乙基或三氟乙基。
8.根据权利要求1至7中任一项所述的化合物,其中r4为甲氧基乙基。
9.根据权利要求1至8中任一项所述的化合物,其中r4为
10.根据权利要求1至9中任一项所述的化合物,其中r5为吗啉基或甲基哌嗪基。
11.根据权利要求1至9中任一项所述的化合物,其中a1为其中键“a”连接到吲哚环。
12.根据权利要求1或2所述的化合物,其中a2为二甲基亚甲基。
13.根据权利要求1或2所述的化合物,其中
14.根据权利要求13所述的化合物,其中
15.一种化合物,其选自:
16.一种具有的结构的化合物rm461-a,或其药用盐。
17.一种用于制备根据权利要求1至15中任一项所述的化合物的方法,所述方法包括以下步骤中的任一者:
18.根据权利要求1至16中任一项所述的化合物或药用盐,其用作治疗活性物质。
19.一种药物组合物,其包含根据权利要求1至16中任一项所述的化合物以及治疗惰性载体。
20.根据权利要求1至16中任一项所述的化合物用于治疗kras g12c蛋白相关疾病的用途。
21.根据权利要求1至16中任一项所述的化合物用于治疗krasg12c、g12d和g12v蛋白相关疾病的用途。
22.根据权利要求1至16中任一项所述的化合物用于抑制ras与下游效应子的相互作用的用途,其中所述下游效应子为raf和pi3k。
23.根据权利要求1至16中任一项所述的化合物用于抑制传播的致癌mapk和pi3k信号传导的用途。
24.根据权利要求1至16中任一项所述的化合物用于治疗或预防kras突变驱动的癌症的用途,其中所述癌症选自胰腺癌、结直肠癌、肺癌、食道癌、胆囊癌、黑素瘤、卵巢癌和子宫内膜癌。
25.根据权利要求1至16中任一项所述的化合物用于治疗或预防kras突变驱动的癌症的用途,其中所述癌症选自胰腺腺癌、结直肠癌和非小细胞肺癌。
26.根据权利要求1至16中任一项所述的化合物或药用盐,其用于治疗或预防kras突变驱动的癌症,其中所述癌症选自胰腺腺癌、结直肠癌和非小细胞肺癌。
27.根据权利要求1至16中任一项所述的化合物用于制备药物的用途,所述药物用于治疗或预防kras突变驱动的癌症,其中所述癌症选自胰腺腺癌、结直肠癌和非小细胞肺癌。
28.一种用于治疗或预防kras突变驱动的癌症的方法,其中所述癌症选自胰腺腺癌、结直肠癌和非小细胞肺癌,所述方法包括施用治疗有效量的如权利要求1至16中任一项中所定义的化合物。
29.根据权利要求1至16中任一项所述的化合物或药用盐,其根据权利要求17所述的方法制造。
30.如前所述的本发明。
技术总结
本发明涉及式(I)化合物,其中R1至R5以及A1至A3如本文所述,及其药用盐;以及包含所述化合物的组合物和使用所述化合物的方法。
技术研发人员:陈建国,刘海侠,裘虹霞,沈宏,阎志鹏,杨北猛,姚向瑜,张卫星,赵丹,朱伟
受保护的技术使用者:豪夫迈·罗氏有限公司
技术研发日:
技术公布日:2024/3/12
- 还没有人留言评论。精彩留言会获得点赞!