一种提高制备六苄基六氮杂异伍兹烷得率的方法
本发明属于含能材料,涉及一种提高制备六苄基六氮杂异伍兹烷得率的方法。
背景技术:
1、六苄基六氮杂异伍兹烷(hbiw)为笼状结构,含能低,由于存在氨基,在酸性条件中极不稳定,容易被破坏,而且苄基的芳香环比笼体中的叔胺硝化竞争能力更强,因此不能直接进行硝解反应,可以通过多种反应制备硝化反应中间体,是合成含能材料的重要原料。
2、cl-20是当今含能材料领域的热点,虽然有各类的cl-20合成前体,但是直链缩合和苯环上带取代基的胺与醛反应后得到产物的收率低,副产物多,而且后续步骤难以进行,因此hbiw仍是合成cl-20的主要前体。作为后续反应的一环,其工业化也是我们重点研究的目标。
3、目前hbiw的合成大多采用一锅法,采用甲酸作为催化剂,反应过程复杂,副产物较多,小试收率为70-80%,工业放大收率55-65%。重复文献方法,发现小试合成的稳定收率仅有73%,因此需要对其合成工艺进行探究,寻找适宜的反应条件。
4、尽管有很多文献对制备hbiw进行过报道,但大多数这些方法都存在以下一个或多个缺点:反应时间长,产物收率低,反应成本高和副反应多、后处理困难等。这些问题一直没有得到解决。
技术实现思路
1、本发明的目的在于提供一种提高制备hbiw得率的方法。
2、为达到上述目的,本发明的技术方案是:利用釜式连续搅拌反应器合成hbiw的方法。
3、本发明所述的合成方法包括以下具体步骤:
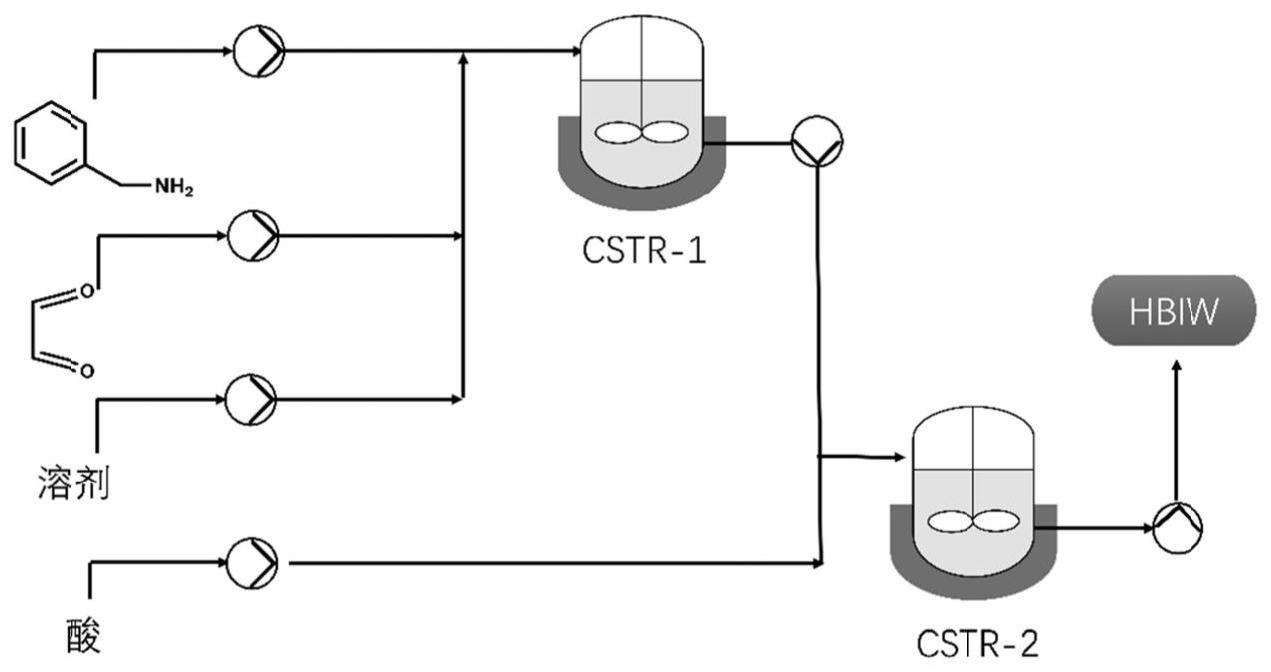
4、步骤1)将乙腈和水的混合溶液、苄胺与乙二醛通过注射泵同时送入第一连续搅拌釜式反应器(cstr-1)进行混合反应,反应0.5-1.5h,cstr-1的反应温度控制在10-30℃。
5、步骤2)将步骤(1)反应后溶液中通过蠕动泵转移到第二连续搅拌釜式反应器(cstr-2),同时将酸通过注射泵送入cstr-2中,继续反应0.5-1.5h,cstr-2中的温度控制在10-30℃,继而得到目标产物。
6、步骤3)将步骤(2)得到的粗产物,用溶剂进行重结晶,得到浅黄色针状晶体的步骤。
7、本发明与现有技术相比,显著优点是:本发明采用的是釜式搅拌连续反应器进行hbiw的合成,有效的克服了在合成过程中的时间长,物质混合不均,产率不高的问题,使得反应的产率和安全性均得到改善。有望成为一种新型的制备hbiw的方法。
技术特征:
1.一种提高制备六苄基六氮杂异伍兹烷得率的方法,其特征在于,包括如下步骤:
2.根据权利要求1所述的一种提高制备六苄基六氮杂异伍兹烷得率的方法,其特征在于,步骤(1)中,以摩尔比计,苄胺:乙二醛=2:0.5-2:1。
3.根据权利要求1所述的一种提高制备六苄基六氮杂异伍兹烷得率的方法,其特征在于,步骤(1)中,乙腈和水的混合溶液的溶剂比为10:1-40:1。
4.根据权利要求1所述的一种提高制备六苄基六氮杂异伍兹烷得率的方法,其特征在于,步骤(1)中,苄胺和乙腈溶剂比为6:1-10:1。
5.根据权利要求1所述的一种提高制备六苄基六氮杂异伍兹烷得率的方法,其特征在于,步骤(1)中,将苄胺、乙二醛、有机溶剂同时通过注射泵送入第一连续搅拌釜式反应器,反应时间为0.5-1.5小时。
6.根据权利要求1所述的一种提高制备六苄基六氮杂异伍兹烷得率的方法,其特征在于,步骤(1)中,第一连续搅拌釜式反应器中的反应温度控制在10-30℃。
7.根据权利要求1所述的一种提高制备六苄基六氮杂异伍兹烷得率的方法,其特征在于,步骤(2)中,所用酸选自甲酸、乙酸、氨基磺酸,对甲苯磺酸、苯甲酸中的任意一种。
8.根据权利要求1所述的一种提高制备六苄基六氮杂异伍兹烷得率的方法,其特征在于,步骤(2)中,酸的添加量与苄胺的摩尔量的比为0.03:1-0.1:1。
9.根据权利要求1所述的一种提高制备六苄基六氮杂异伍兹烷得率的方法,其特征在于,步骤(2)中,将酸和步骤(1)反应后溶液同时送入第二连续搅拌釜式反应器中,反应时间为0.5-2h。
10.根据权利要求1所述的一种提高制备六苄基六氮杂异伍兹烷得率的方法,其特征在于,步骤(2)中,第二连续搅拌釜式反应器中的反应温度控制在10-30℃。
技术总结
本发明公开了一种采用连续搅拌釜式反应器(CSTR)制备六苄基六氮杂异伍兹烷(HBIW)的方法,该方法能够提高HBIW的得率,显著缩短反应时间。本发明首先将乙腈和水的混合溶液、苄胺和乙二醛同时通过注射泵送入CSTR‑1进行混合反应,反应40min后,将反应后溶液通过蠕动泵送入CSTR‑2,同时将甲酸通过注射泵送入CSTR‑2,混合反应40min,即可得到产物HBIW,该过程使得制备HBIW的得率提高到了87.56%。本发明采CSTR具有高传质和高传热的基本特征,以及强比表面积和更高的安全性,反应过程物料高度混合,更加高效环保。同时该方法具有反应工艺简单、易于大规模连续生产、收率高等优点。
技术研发人员:林秋汉,朱咪咪,张立南,马慧朝,陆明
受保护的技术使用者:南京理工大学
技术研发日:
技术公布日:2024/1/15
- 还没有人留言评论。精彩留言会获得点赞!