双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的制备方法与流程
本发明属于有机金属化合物合成,具体涉及双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的制备方法。
背景技术:
1、氧化镁(mgo)广泛用于多层电子/光子器件的绝缘和缓冲层,由于其具有非常大的带隙(7.2 ev),优异的热稳定性(熔点2900℃)和电绝缘性能(介电常数9.8),且具有简单的立方岩盐晶体结构,易形成具有高度mgo(100)取向的织化微结构的薄膜。利用离子束辅助沉积(ibad)技术成功地制备了具有高度双轴结构的mgo薄膜用作ybco超导的模板/缓冲层涂层,以促进模板工艺,阻止电串扰,并尽量减少界面扩散和晶格不匹配。mgo织构薄膜也常用于提高磁性存储介质中薄膜的结晶度,同时降低衬底成本。单晶mgo晶片是优异的外延薄衬底,由于其良好的表面条件,薄膜生长时从衬底到薄膜传播所需的微结构织构。此外,mgo薄膜具有优异的耐火性能和低溅射率,在改善交流等离子体显示面板的放电特性和面板寿命缺陷方面发挥了重要的保护层作用。
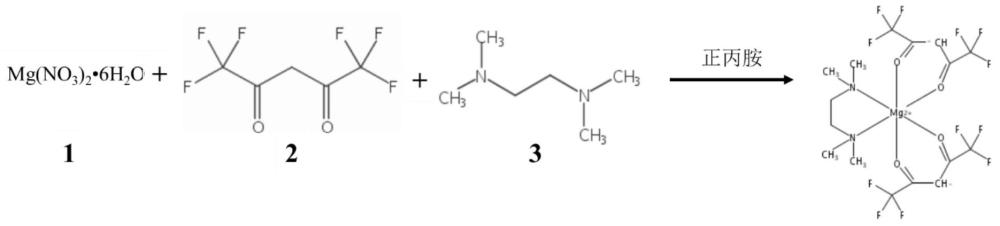
2、迄今为止,mgo薄膜的沉积技术有几种,包括溶胶凝胶、溅射、脉冲激光沉积(pld)、离子束辅助沉积(ibad)和化学气相沉积(cvd)。在这些技术中,金属有机化学气相沉积(mocvd)具有设备相对简单、适合大规模生产、保形覆盖范围广、沉积材料适用范围广等优点。生长在非晶玻璃衬底上的mgo薄膜通常是多晶材料。通过mocvd成功生长薄膜的关键因素是有效的金属有机前驱体的可用性,而高纯度的双(六氟乙酰丙酮)(四甲基乙二胺)镁可作为原子层沉积法制备mgo薄膜的前驱体源材料。
3、文献chem. mater., vol. 17, no. 23, 2005 5699,设计了双(六氟乙酰丙酮)(四甲基乙二胺)镁前驱体,并通过饱和配位来提高前驱体的挥发性和热稳定性,解决了当前mg前驱体的不足。但该文献中镁前驱体的合成方法仍存在收率和纯度较低的问题,亟需解决。
技术实现思路
1、针对上述技术问题,本申请提供双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的制备方法。
2、为实现上述目的,本申请提出如下技术方案:
3、本申请提供双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的制备方法,包括:
4、(1)向六氟乙酰丙酮水溶液中滴加正丙胺,待正丙胺滴加完毕后,继续反应,得到溶液a;
5、(2)向硝酸镁水溶液中滴加溶液a,待溶液a滴加完毕后,继续反应,得到双(六氟乙酰丙酮)镁溶液;
6、(3)向双(六氟乙酰丙酮)镁溶液中滴加n,n,n',n'-四甲基乙二胺,待滴加完毕后,继续反应,反应后经抽滤得到滤渣,滤渣经干燥,得到粗产物;
7、(4)将粗产物先于50~60℃下进行减压蒸馏直至无低沸点杂质蒸出,然后升温至70~80℃,升华得到目标产物双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物。
8、进一步地,所述六氟乙酰丙酮、硝酸镁和n,n,n',n'-四甲基乙二胺的摩尔比为2~2.2:1:1~1.2。
9、进一步地,所述正丙胺与所述六氟乙酰丙酮水溶液中的六氟乙酰丙酮的摩尔比为1~1.3:1。
10、进一步地,步骤(4)中,所述减压蒸馏的压力为1~1.5mtorr。
11、进一步地,步骤(4)中,所述升华的压力为1~1.5mtorr。
12、进一步地,步骤(3)中,所述继续反应的温度为15~35℃。
13、进一步地,步骤(3)中,所述继续反应在搅拌条件下进行。
14、进一步地,步骤(3)中,所述滴加的速度为1~5滴/s;滴加n,n,n',n'-四甲基乙二胺时对所述双(六氟乙酰丙酮)镁溶液进行搅拌;滴加n,n,n',n'-四甲基乙二胺时所述双(六氟乙酰丙酮)镁溶液的温度为-5~5℃。
15、进一步地,步骤(1)中,所述滴加的速度为1~5滴/s;滴加正丙胺时对所述六氟乙酰丙酮水溶液进行搅拌;滴加正丙胺时所述六氟乙酰丙酮水溶液的温度为-5~5℃。
16、进一步地,步骤(1)中,所述继续反应的温度为15~35℃。
17、进一步地,步骤(1)中,所述继续反应在搅拌条件下进行。
18、进一步地,步骤(1)中,所述六氟乙酰丙酮水溶液的浓度为100~160g/l。
19、进一步地,步骤(2)中,所述硝酸镁水溶液的浓度为30~70g/l。
20、进一步地,步骤(2)中,所述溶液a的滴加速度为1~5滴/s;滴加溶液a时对所述硝酸镁水溶液进行搅拌;滴加溶液a时所述硝酸镁水溶液的温度为-5~5℃。
21、进一步地,步骤(2)中,所述继续反应的温度为-5~5℃。
22、进一步地,步骤(2)中,所述继续反应在搅拌条件下进行。
23、进一步地,步骤(4)中,所述双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的有机含量为99.99%以上;所述双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的无机纯度为6n。
24、与现有技术相比,上述技术方案之一或多个技术方案能达到至少以下有益效果之一:
25、上述制备方法高效实现了高纯双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的制备,且该制备方法的产物收率高、纯度高。
26、上述制备方法利用单一水作为溶剂,减少了有机溶剂的使用,进而规避了产物溶于有机溶剂中而造成损失。
27、上述制备方法通过优化原料的加料、反应顺序,有效减少了副产物的生成,进而提高了各步收率。
28、使用减压升华的方法进行提纯,无需额外添加试剂,不仅降低了成本,而且简化了提纯步骤,提高了效率,更有利于工业化生产。
技术特征:
1.双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的制备方法,其特征在于,包括:
2.如权利要求1所述的双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的制备方法,其特征在于,所述六氟乙酰丙酮、硝酸镁和n,n,n',n'-四甲基乙二胺的摩尔比为2~2.2:1:1~1.2。
3.如权利要求1所述的双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的制备方法,其特征在于,所述正丙胺与所述六氟乙酰丙酮水溶液中的六氟乙酰丙酮的摩尔比为1~1.3:1。
4.如权利要求1所述的双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的制备方法,其特征在于,步骤(4)中,所述减压蒸馏的压力为1~1.5mtorr;
5.如权利要求1所述的双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的制备方法,其特征在于,步骤(3)中,所述继续反应的温度为15~35℃;所述继续反应在搅拌条件下进行。
6.如权利要求1所述的双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的制备方法,其特征在于,步骤(3)中,所述滴加的速度为1~5滴/s;滴加n,n,n',n'-四甲基乙二胺时对所述双(六氟乙酰丙酮)镁溶液进行搅拌;滴加n,n,n',n'-四甲基乙二胺时所述双(六氟乙酰丙酮)镁溶液的温度为-5~5℃。
7.如权利要求1所述的双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的制备方法,其特征在于,步骤(1)中,所述滴加的速度为1~5滴/s;滴加正丙胺时对所述六氟乙酰丙酮水溶液进行搅拌;滴加正丙胺时所述六氟乙酰丙酮水溶液的温度为-5~5℃;
8.如权利要求1~7任意一项所述的双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的制备方法,其特征在于,步骤(1)中,所述六氟乙酰丙酮水溶液的浓度为100~160g/l;
9.如权利要求1所述的双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的制备方法,其特征在于,步骤(2)中,所述溶液a的滴加速度为1~5滴/s;滴加溶液a时对所述硝酸镁水溶液进行搅拌;滴加溶液a时所述硝酸镁水溶液的温度为-5~5℃;
10.如权利要求1~7任意一项所述的双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的制备方法,其特征在于,步骤(4)中,所述双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的有机含量为99.99%以上;所述双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的无机纯度为6n。
技术总结
本发明提供双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的制备方法,包括:向六氟乙酰丙酮水溶液中滴加正丙胺,待滴加完毕后继续反应,得到溶液A;向硝酸镁水溶液中滴加溶液A,待溶液A滴加完毕后,继续反应,得到双(六氟乙酰丙酮)镁溶液;向双(六氟乙酰丙酮)镁溶液中滴加N,N,N',N'‑四甲基乙二胺,滴加完毕后,继续反应,反应后经抽滤得到滤渣,滤渣经干燥,得到粗产物;将粗产物先进行减压蒸馏直至无低沸点杂质蒸出,然后升温,升华得到目标产物双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物。上述制备方法高效实现了高纯双(六氟乙酰丙酮)(四甲基乙二胺)镁配合物的制备,且所得产物收率高、纯度高。
技术研发人员:周锦荣,翁宇轩,潘唐纳,徐成,曾翼俊,冯威
受保护的技术使用者:广东先导微电子科技有限公司
技术研发日:
技术公布日:2024/8/26
- 还没有人留言评论。精彩留言会获得点赞!