一种吲哚美辛酯类衍生物的制备方法
本发明涉及一种吲哚美辛衍生物的制备方法,尤其涉及一种吲哚美辛酯类衍生物的制备方法。
背景技术:
1、吲哚美辛是临床上常见的一种非甾体类抗炎药,此类药物大多为非特异性的环氧化酶(cox)抑制剂,即:可同时抑制环氧合酶-1(cyclooxygenase-1,cox-1)和环氧合酶-2(cyclooxygenase-2,cox-2)。进一步地研究表明,对cox-2抑制作用越大,其消炎、止痛效果越显著;对cox-1的抑制作用越强,则不良反应越大(drugs,1997,53,563-582)。
2、现有研究发现,将吲哚美辛上羧酸基团酯化后得到的吲哚美辛酯类衍生物对cox-2具有高效的特异性抑制作用(proc.natl.acad.sci.u.s.a.2000,97,925-930)。由此,对于吲哚美辛酯类衍生物的制备方法相继被报道出来,主要有如下两种方法:
3、
4、(a)方法需要加入1,3-二环己基碳亚胺(dcc)和4-二甲氨基吡啶(dmap)促进吲哚美辛与醇类化合物进行缩合,但是该类反应最大的缺点在于反应物的副产物二环己基脲以常用的纯化方法很难将其完全除去,影响产物的纯度。(b)方法需要使用六甲基磷酰三胺(hmpa),此化合物具有潜在致癌物,与皮肤接触24小时能引起炎症,安全隐患极大。因此,开发简洁、安全、高效制备吲哚美辛酯类衍生物的方法变得尤为重要。
技术实现思路
1、发明目的:本发明的目的是提供一种吲哚美辛酯类衍生物的制备方法,解决现有制备方法存在的副产物难以去除和反应试剂毒性较大的问题。
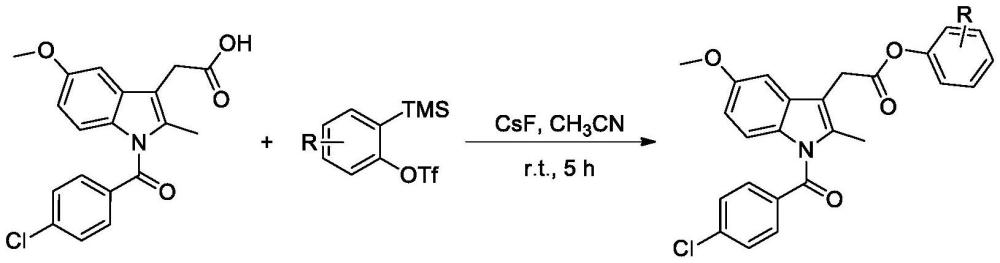
2、技术方案:本发明所述的一种吲哚美辛酯类衍生物的制备方法,包括如下步骤:
3、
4、其中,r1选自h、卤素、c1-3烷基、c1-3烷氧基、环烷烃或芳香烃类基团;r2选自h或卤素,tms为三甲基硅基团,otf为三氟甲磺酸酯基团。
5、化合物iii的结构式如下:
6、
7、其中,r1、r2可处于苯环的任意可取代位置。
8、优选地,所述碱为氟化铯,溶剂为乙腈、三氯甲烷、乙酸乙酯、四氢呋喃中的至少一种。
9、优选地,所述化合物ii与化合物iii的摩尔比为1:1-2。
10、优选地,所述化合物ii与碱的摩尔比为1:1-3。
11、优选地,化合物ii和化合物iii的反应条件为20-30℃反应4-10小时。
12、优选地,反应后还包括如下步骤:粗产物用水洗涤,乙酸乙酯萃取,减压旋干后,用体积比为1:10-20的乙酸乙酯:石油醚为洗脱剂进行柱层析分离纯化。
13、优选地,所述化合物ii在溶剂中的浓度为0.1-0.25mol/l。
14、优选地,所述化合物iii为如下化合物中的至少一种:
15、
16、由上述化合物iii对应制得的化合物i为如下任一种:
17、
18、本发明的制备反应机理如下:
19、
20、2-(三甲基硅基)苯基三氟甲磺酸酯类化合物在氟化铯中的氟离子作用下离去三甲基硅基(-tms)基团和三氟甲磺酸基(-otf)基团便能原位生成苯炔中间体,随后被吲哚美辛上的羧基氧原子以亲核进攻的形式所捕获,最终得到吲哚美辛酯类衍生物。
21、有益效果:与现有技术相比,本发明具有如下显著优点:本发明合成方法简单,无需外加缩合剂或金属催化剂,为开发吲哚美辛酯类衍生物提供了一种全新的途径。
技术特征:
1.一种吲哚美辛酯类衍生物的制备方法,其特征在于,包括如下步骤:
2.根据权利要求1所述吲哚美辛酯类衍生物的制备方法,其特征在于,所述碱为氟化铯,溶剂为乙腈、三氯甲烷、乙酸乙酯、四氢呋喃中的至少一种。
3.根据权利要求1所述吲哚美辛酯类衍生物的制备方法,其特征在于,所述化合物ii与化合物iii的摩尔比为1:1-2。
4.根据权利要求1所述吲哚美辛酯类衍生物的制备方法,其特征在于,所述化合物ii与碱的摩尔比为1:1-3。
5.根据权利要求1所述吲哚美辛酯类衍生物的制备方法,其特征在于,化合物ii和化合物iii的反应条件为20-30℃反应4-10小时。
6.根据权利要求1所述吲哚美辛酯类衍生物的制备方法,其特征在于,反应后还包括如下步骤:粗产物用水洗涤,乙酸乙酯萃取,减压旋干后,用体积比为1:10-20的乙酸乙酯:石油醚为洗脱剂进行柱层析分离纯化。
7.根据权利要求1所述吲哚美辛酯类衍生物的制备方法,其特征在于,所述化合物ii在溶剂中的浓度为0.1-0.25mol/l。
8.根据权利要求1所述吲哚美辛酯类衍生物的制备方法,其特征在于,所述化合物iii为如下化合物中的至少一种:
技术总结
本发明公开了一种吲哚美辛酯类衍生物的制备方法。吲哚美辛酯类衍生物如式(I)所示。该衍生物的制备方法是以2‑(三甲基硅基)苯基三氟甲磺酸酯类化合物为苯炔前体,氟化铯作碱,实现无需外加缩合剂或金属催化剂条件下和吲哚美辛反应,该方法操作简单、步骤简洁,为有重要价值的吲哚美辛酯类衍生物的制备提供了安全、高效、实用的合成方法。本发明制得的吲哚美辛酯类衍生物有望用于制备COX‑2抑制剂或非甾体类抗炎药物。
技术研发人员:姚亮亮,张远,丁晶晶,刘迪,吴豪
受保护的技术使用者:滁州学院
技术研发日:
技术公布日:2024/12/10
- 还没有人留言评论。精彩留言会获得点赞!