本发明涉及生物检测技术领域,特别涉及一种基于高通量测序技术的宏转录组数据分析方法。
背景技术:
宏转录组学(metatranscriptomics)的研究对象是微生物组mrna,在获取微生物组总rna并去除rrna之后,反转录为cdna,并构建合适长度的插入片段文库,对这些文库进行双端(paired-end,pe)高通量测序,从而能精确定量整个菌群中具有活性的物种精细组成及其对应功能的表达水平,进而锁定菌群中的关键生物标记物、阐明其生物学意义。
技术实现要素:
本发明所要解决的技术问题在于提供一种基于高通量测序技术的宏转录组数据分析方法。
本发明所要解决的技术问题可以通过以下技术方案来实现:
一种基于高通量测序技术的宏转录组数据分析方法,具体包括如下步骤:
(1)对高通量测序下机的双端序列原始数据进行质量筛查,获取可用于下游宏转录组学分析的高质量数据集;
(2)对高质量序列进行核糖体rna序列预测和剔除,得到mrna的转录本序列集;
(3)对每个样本分别进行宏转录组序列拼接组装,构建宏转录组contigs和scaffolds序列集,并进行基因预测,获得非冗余蛋白序列集;
(4)对蛋白序列用多种常用数据库进行功能注释,获得各等级的功能类群丰度谱,并进行差异比较分析、代谢通路富集分析、聚类分析;
(5)对基因序列进行物种注释,获得种以及种以下精细水平的物种组成谱,并进行差异比较分析、聚类分析、物种组成丰富度和均匀度分析和关联网络分析;
(6)基于上述获得的功能丰度谱和物种组成谱,可以进一步对宏转录组样本进行alpha和beta多样性分析,进而依靠多种多变量统计学方法筛选得到宏基因组中的关键生物标记物;
(7)通过多种数据可视化和交互式工具,绘制二维/三维图表,全方位、客观地呈现以上分析结果;
(8)根据样本来源,选择特定的功能数据库进行注释分析。
由于采用了如上的技术方案,本发明具有如下特点:
(1)直接对菌群样本中活性表达的基因片段进行测序,真正实现对活性物种和表达功能的精确定量;
(2)多种功能注释数据库可选,根据研究需求选择kegg/eggnog/cazy/nr/swiss-prot/go/vfdb/card等数据库,最优化宏转录组的活性功能代谢谱注释;
(3)通过微生物基因信息精确识别物种来源,获取种以及种以下水平的“高分辨率”活性物种精细组成谱;
(4)通过多种多变量统计分析和机器学习方法,系统、深入地挖掘宏转录组大数据中差异相关的活性物种和对应功能,从而精确识别关键的活性生物标记物。
附图说明
图1为本发明一种基于高通量测序技术的宏转录组数据分析方法的流程示意图。
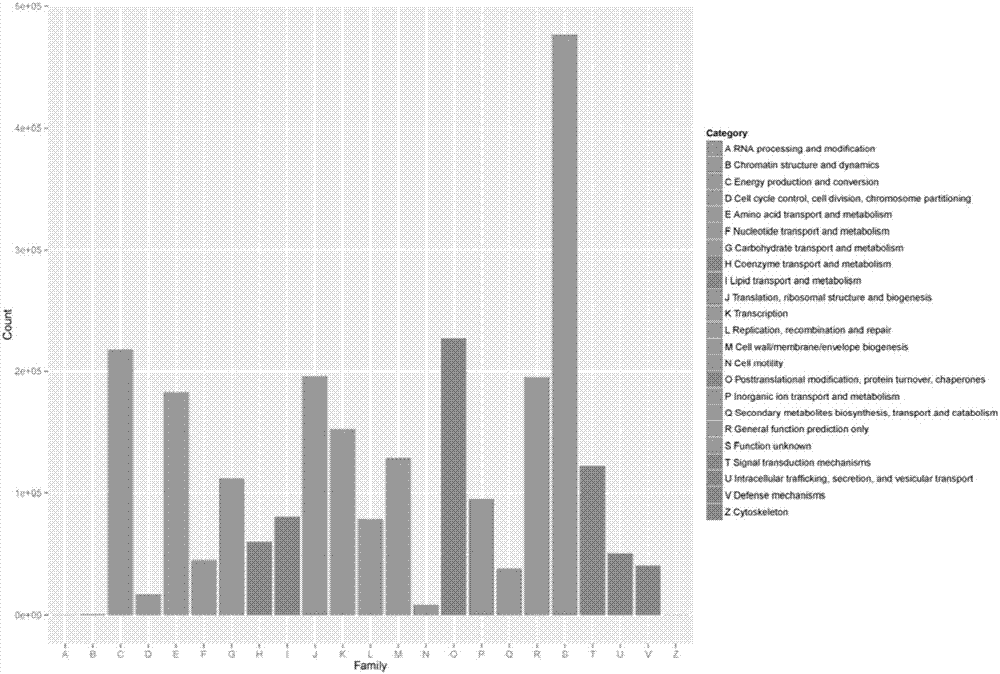
图2为本发明的eggnog功能类群的注释结果统计图。图中,横座标对应eggnog的25个基因功能大类,每一大类以一个英语大写字母代表,纵坐标为注释到对应分类的eggnog功能类群数量。
图3本发明的unigene差异表达ma图。图中,横坐标表明每个unigene在两样本(组)中的平均表达强度(即a值,a=[log2(case)+log2(control)]/2,case和control分别代表该unigene在两样本(组)中的表达量),横坐标值越大,对应unigene的平均表达强度越强。纵坐标为每个unigene在两样本(组)间的表达量倍数差异对数值(即m值,m=log2(control/case)),纵坐标对数值越大,对应unigene在control样本(组)中的表达量越高,而在case样本(组)中的表达量越低;对数值越小,对应unigene在case样本(组)中的表达量越高,而在control样本(组)中的表达量越低。在两样本(组)中差异表达的unigene在图上以红色圆点表示,表达量无差异的unigene以青色圆点表示。
图4为本发明的显示效果图。基于各样本在kegg功能数据库中注释得到的ko功能类群的相对表达量分布表,可以分析每个样本(组)所富集(即表达量显著上调)的ko,并通过统计检验评价差异是否显著。代谢通路富集效果的展示形式将根据所选择的功能类别有所不同。
图5为本发明的phi数据库注释结果统计图。图中,横座标对应phi的9个基因大类,纵坐标为注释到对应分类的基因数量。
具体实施方式
参见图1,图中给出的一种基于高通量测序技术的宏转录组数据分析方法,具体包括如下步骤:
(1)对高通量测序下机的双端序列原始数据进行质量筛查,获取可用于下游宏转录组学分析的高质量数据集;
(2)对高质量序列进行核糖体rna序列预测和剔除,得到mrna的转录本序列集;
(3)对每个样本分别进行宏转录组序列拼接组装,构建宏转录组contigs和scaffolds序列集,并进行基因预测,获得非冗余蛋白序列集;
(4)对蛋白序列用多种常用数据库进行功能注释,获得各等级的功能类群丰度谱,并进行差异比较分析、代谢通路富集分析、聚类分析(参见图2、图3);
(5)对基因序列进行物种注释,获得种以及种以下精细水平的物种组成谱,并进行差异比较分析、聚类分析、物种组成丰富度和均匀度分析和关联网络分析(参见图4);
(6)基于上述获得的功能丰度谱和物种组成谱,可以进一步对宏转录组样本进行alpha和beta多样性分析,进而依靠多种多变量统计学方法筛选得到宏基因组中的关键生物标记物(参见图5);
(7)通过多种数据可视化和交互式工具,绘制二维/三维图表,全方位、客观地呈现以上分析结果;
(8)根据样本来源,选择特定的功能数据库进行注释分析。
技术特征:技术总结本发明公开的一种基于高通量测序技术的宏转录组数据分析方法,包括如下步骤:(1)获得高质量数据集;(2)得到mRNA的转录本序列集;(3)获得非冗余蛋白序列集;(4)获得各等级的功能类群丰度谱并进行分析;(5)对基因序列进行物种注释,获得种以及种以下精细水平的物种组成谱,并进行分析;(6)基于上述获得的功能丰度谱和物种组成谱,进一步对宏转录组样本进行Alpha和Beta多样性分析,进而依靠多种多变量统计学方法筛选得到宏基因组中的关键生物标记物;(7)通过多种数据可视化和交互式工具,绘制二维/三维图表,全方位、客观地呈现以上分析结果;(8)根据样本来源,选择特定的功能数据库进行注释分析。
技术研发人员:薛正晟;杨洋;姜丽荣;孙子奎
受保护的技术使用者:上海派森诺生物科技股份有限公司
技术研发日:2017.08.21
技术公布日:2018.01.19