一种基于分子动力学的模拟石墨烯吸附角蛋白过程的方法与流程
本发明属于石墨烯吸附蛋白质研究,具体涉及一种基于分子动力学的模拟石墨烯吸附角蛋白过程的方法。
背景技术:
1、角蛋白作为人体皮肤角质层中含量最多的一种蛋白质,可以维持皮肤的结构形态并保护机体不受外界环境的伤害。石墨烯作为二维纳米材料,具有优异的力学性能,导电性能和热力学性能,已经被广泛用在柔性皮肤、药物载体等领域。温度作为一种最普遍的环境因素,对石墨烯的各种理化性能具有重要影响。因此对温度影响石墨烯和角蛋白之间的吸附过程研究得到越来越多的关注。
2、石墨烯与蛋白质之间只存在范德华相互作用力,在吸附角蛋白的过程中,会造成角蛋白二级结构的不断变化,相应的,角蛋白的各项力学性能和热稳定性等也会随之改变。石墨烯属于纳米尺度材料,传统实验仪器,如原子力显微镜,红外光谱仪、透射电子显微镜和扫描电子显微镜等价格昂贵,操作复杂,受实验环境和操作水平的影响较大,并且传统的实验仪器获取的是静态数据,以上因素都严重限制了对石墨烯吸附角蛋白过程的进一步研究。
3、分子动力学模拟是以计算机为核心的科学研究方法,相比于传统的实验手段,其更加的高效和便宜,并且能在不同时空尺度动态模拟整个反应过程,因此,分子动力学模拟已经被广泛应用在材料和生物医学等行业。但是,到目前为止,通过分子动力学模拟研究温度影响石墨烯吸附含螺旋结构域的角蛋白的研究尚未见相关报道。
技术实现思路
1、针对现有技术的不足,本发明提供一种基于分子动力学的模拟石墨烯吸附角蛋白过程的方法,该方法相比于传统实验方案,具有操作简单、成本低、精度高等优点。
2、本发明是通过以下技术方案实现的:
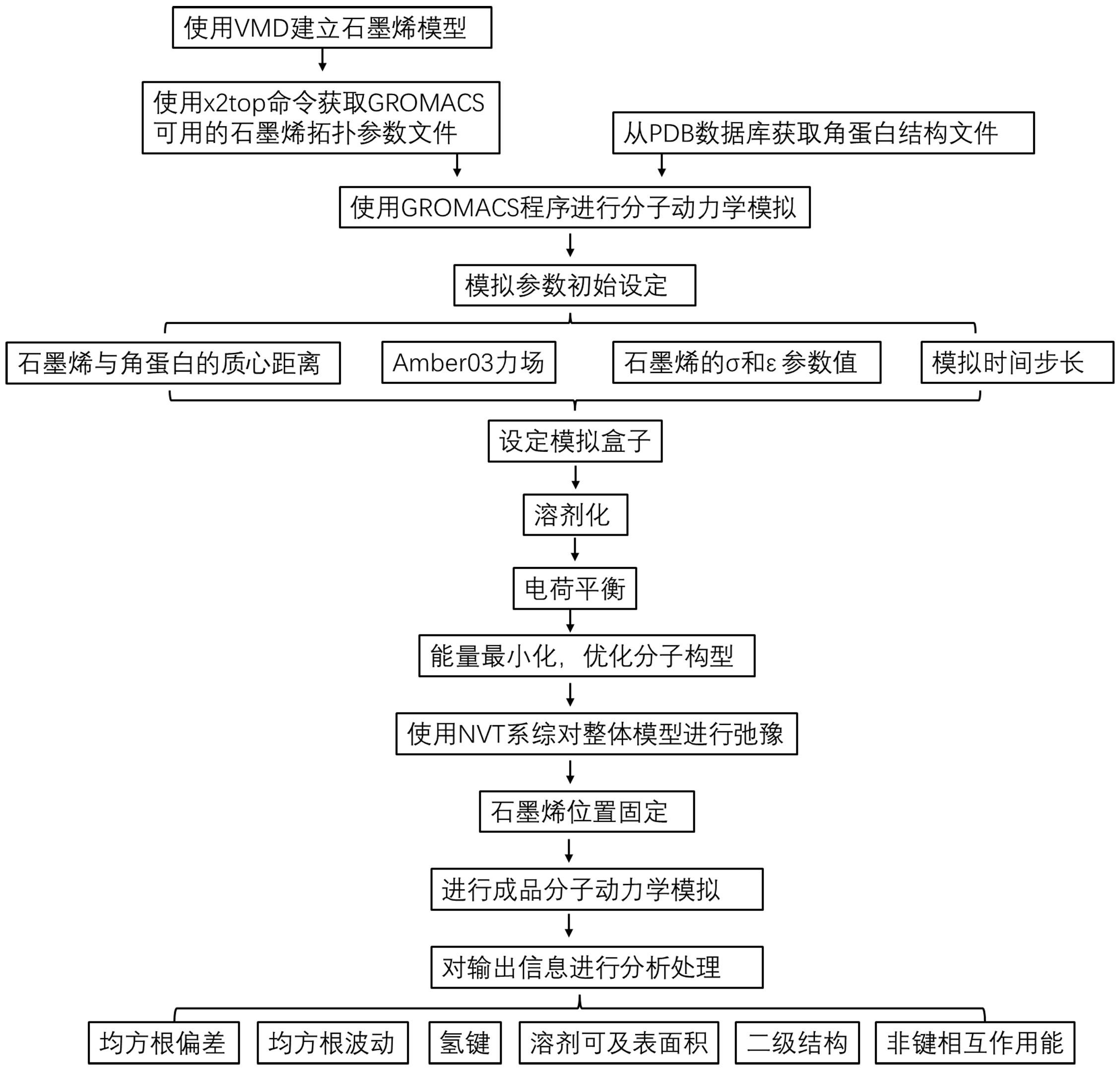
3、一种基于分子动力学的模拟石墨烯吸附角蛋白过程的方法,包括以下步骤:
4、步骤1)由vmd软件中的nanotube builder模块构建石墨烯模型,在amber03分子力场的n2t文件中编写石墨烯的成键参数,通过gromacs软件中的x2top命令将建立好的石墨烯模型的坐标文件转换为gromacs可识别的拓扑文件;
5、步骤2)下载角蛋白结构坐标文件,由vmd软件设定石墨烯与角蛋白的模拟初始位置文件,再将两者的结构文件合并为初始结构坐标输入文件,得到初始结构坐标文件a;
6、步骤3)通过gromacs软件中的editconf命令,将步骤2)所得的初始结构坐标文件a置于虚拟盒子中心,且初始结构与盒子边界最小距离不低于1 nm,得到新的结构坐标文件b;
7、步骤4)通过gromacs软件中的solvate命令,将步骤3)的结构坐标文件b填满水溶剂,得到新的结构坐标文件c;
8、步骤5)通过gromacs软件中的genion命令,将步骤4)的结构坐标文件c进行电荷平衡,得到新的结构坐标文件d;
9、步骤6)对步骤5)的结构坐标文件d进行能量最小化,排除不合理结构,得到新的结构坐标文件e;
10、步骤7)经步骤6)结构优化后,使用freeze命令对步骤6)的结构坐标文件e中的石墨烯组固定;
11、步骤8)对步骤6)的结构坐标文件e采用nvt系综进行弛豫,弛豫过程中的控温方式选用velocity-rescale方法,0 ps、100 ps、200 ps的退火温度点分别设定为0、0.5t、t,随后将温度保持在310 k;设置模拟时间步长为1 fs,得到弛豫后的新结构坐标文件f;
12、步骤9)对步骤8)得到的结构坐标文件f进行成品动力学模拟,此过程中控温方式选用velocity-rescale方法,0 ps、100 ps、200 ps的退火温度点分别设定为0、0.5t、t,随后将温度保持在310 k;设置模拟时间步长为2 fs,得到最终的结构坐标文件g,完成对石墨烯吸附角蛋白过程的分子动力学模拟。
13、优选地,步骤8)和步骤9)中所述t的温度范围为277~315 k。
14、优选地,步骤9)得到的结构坐标文件g可以被进一步分析处理,通过gromacs软件中的rms命令,可以计算角蛋白在吸附过程中的均方根偏差数据。
15、优选地,步骤9)得到的结构坐标文件g可以被进一步分析处理,通过gromacs软件中的rmsf命令,可以计算角蛋白在吸附过程中的均方根波动数据。
16、优选地,步骤9)得到的结构坐标文件g可以被进一步分析处理,通过gromacs软件中的hbond命令,可以计算角蛋白在吸附过程中的氢键变化数据。
17、优选地,步骤9)得到的结构坐标文件g可以被进一步分析处理,通过gromacs软件中的do_dssp命令,可以计算角蛋白在吸附过程中的二级结构变化数据。
18、优选地,步骤9)得到的结构坐标文件g可以被进一步分析处理,通过gromacs软件中的sasa命令,可以计算角蛋白在吸附过程中溶剂可及表面积数据。
19、优选地,步骤9)得到的结构坐标文件g可以被进一步分析处理,通过gromacs软件中的energy命令,可以计算石墨烯吸附角蛋白过程中,两者间的相互作用能变化数据。
20、本发明的有益效果如下:
21、本发明利用分子动力学模拟方法研究石墨烯与角蛋白的吸附过程,并动态分析吸附过程中,角蛋白的二级结构变化情况。具体为采用一种适用于石墨烯分子动力学模拟的拓扑结构参数,并通过合理选择原子间相互作用势函数、设定恰当的分子动力学模拟参数和步骤,可以精确模拟计算石墨烯和角蛋白之间的相互作用力,如范德华力,并能准确仿真角蛋白在吸附过程中的二级结构、氢键数量、骨架波动等参数的变化。本发明的方法相比于传统实验方案,具有操作简单、成本低、精度高等优点,有利于进一步理解石墨烯在柔性皮肤和药物载体等领域对皮肤角蛋白的影响,有助于石墨烯在上述领域的安全性进一步评估,推进石墨烯在上述领域的进一步广泛应用。
技术特征:
1.一种基于分子动力学的模拟石墨烯吸附角蛋白过程的方法,其特征在于,包括以下步骤:
2.根据权利要求1所述的一种基于分子动力学的模拟石墨烯吸附角蛋白过程的方法,其特征在于,步骤8)和步骤9)中所述t的温度范围为277~315 k。
3.根据权利要求1所述的一种基于分子动力学的模拟石墨烯吸附角蛋白过程的方法,其特征在于,步骤9)得到的结构坐标文件g可以被进一步分析处理,通过gromacs软件中的rms命令,可以计算角蛋白在吸附过程中的均方根偏差数据。
4.根据权利要求1所述的一种基于分子动力学的模拟石墨烯吸附角蛋白过程的方法,其特征在于,步骤9)得到的结构坐标文件g可以被进一步分析处理,通过gromacs软件中的rmsf命令,可以计算角蛋白在吸附过程中的均方根波动数据。
5.根据权利要求1所述的一种基于分子动力学的模拟石墨烯吸附角蛋白过程的方法,其特征在于,步骤9)得到的结构坐标文件g可以被进一步分析处理,通过gromacs软件中的hbond命令,可以计算角蛋白在吸附过程中的氢键变化数据。
6.根据权利要求1所述的一种基于分子动力学的模拟石墨烯吸附角蛋白过程的方法,其特征在于,步骤9)得到的结构坐标文件g可以被进一步分析处理,通过gromacs软件中的do_dssp命令,可以计算角蛋白在吸附过程中的二级结构变化数据。
7.根据权利要求1所述的一种基于分子动力学的模拟石墨烯吸附角蛋白过程的方法,其特征在于,步骤9)得到的结构坐标文件g可以被进一步分析处理,通过gromacs软件中的sasa命令,可以计算角蛋白在吸附过程中溶剂可及表面积数据。
8.根据权利要求1所述的一种基于分子动力学的模拟石墨烯吸附角蛋白过程的方法,其特征在于,步骤9)得到的结构坐标文件g可以被进一步分析处理,通过gromacs软件中的energy命令,可以计算石墨烯吸附角蛋白过程中,两者间的相互作用能变化数据。
技术总结
本发明公开了一种基于分子动力学的模拟石墨烯吸附角蛋白过程的方法,采用适用于石墨烯分子动力学模拟的拓扑结构参数,并通过合理选择原子间相互作用势函数、设定恰当的分子动力学模拟参数和步骤,可以精确模拟计算石墨烯和角蛋白之间的相互作用力,如范德华力,并能准确仿真角蛋白在吸附过程中的二级结构、氢键数量、骨架波动等参数的变化。本发明的方法相比于传统实验方案,具有操作简单、成本低、精度高等优点,有利于进一步理解石墨烯在柔性皮肤和药物载体等领域对皮肤角蛋白的影响,有助于石墨烯在上述领域的安全性进一步评估,推进石墨烯在上述领域的进一步广泛应用。
技术研发人员:殷常吉,余蕾,程渊
受保护的技术使用者:苏州工业园区蒙纳士科学技术研究院
技术研发日:
技术公布日:2024/1/14
- 还没有人留言评论。精彩留言会获得点赞!