抗体‑药物缀合物冻干制剂的制作方法
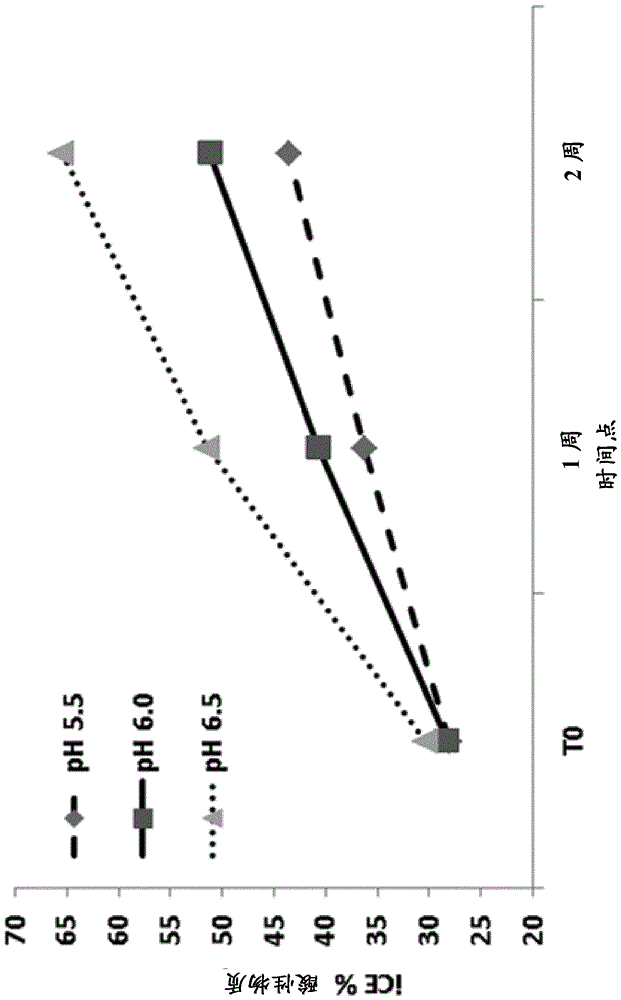
本发明涉及特别适合于抗体药物缀合物(ADC)的冻干制剂,其重构制剂,以及制备这样的冻干和重构制剂的方法和在例如癌症治疗中使用这样的冻干和重构制剂的方法。发明背景ADC是高度有效和特异性的用于治疗癌症和其它病况的药剂,其中抗体部分特异性结合其在靶细胞上的抗原,使得药物可发挥其对靶细胞的细胞毒性或其它治疗效果(任选地在内在化后)。已经描述了数种ADC,其中包括基于抗-组织因子(抗-TF)抗体的ADC(参见例如WO2011157741A2,其通过引用以其整体结合到本文中)。然而,像其它蛋白药物一样,抗体易于降解例如氧化,脱酰胺和片段化以及形成微粒和聚集体。为提供在运输和贮藏期间稳定的ADC药物,因此必须谨慎选择药物制剂中的载体、赋形剂和/或稳定剂。抗体或ADC的长期稳定性也可通过制备冻干或冷冻干燥制剂,使用为该目的而最佳化的赋形剂得到改善。对于抗体或ADC制备物,在专利文献中已经描述了许多这样的制剂,参见例如WO9704801、WO9856418、WO02011753、WO02096457、WO03009817、WO03039485、US8372396、WO2004004639、WO2004016286、WO2004055164、WO2004071439、WO2006014965、WO2006044908和WO2007019232。对于ADC,存在另外的挑战,因为药物缀合本身可降低稳定性和改变抗体的物理化学性质。例如,已报道,药物部分DM1与抗-HER2抗体曲妥单抗的缀合导致抗体的CH2结构域失去稳定性(Wakankar等,2010)。另外,细胞毒性药物通常是疏水的,ADC缀合物作为整体可能比未缀合的抗体更加不溶,因此变得更加易于聚集,形成微粒和表面吸附。通常,抗体和ADC制剂均包括表面活性剂,通常为聚山梨醇酯20或80,以减少聚集和吸附(参见例如,上文引用的专利文献)。例如,brentuximabvedotin(商品名ADCETRIS®)是一种基于与奥里斯他汀(auristatin)衍生物MMAE连接的抗-CD30抗体的ADC,其作为冻干粉末提供,当在水中重构时其包含5mg/mLADC、70mg/mL海藻糖二水合物、5.6mg/mL柠檬酸钠二水合物、0.21mg/mL柠檬酸一水合物和0.20mg/mL聚山梨醇酯80,pH为大约6.6。因此,表面活性剂通常用于药物制剂,并且一般理解为可接受的药物成分。如上所述,在抗体制备和制剂期间表面活性剂通常用于减少聚集体形成(参见例如Vásquez-Rey和Lang,2011,Biotech,Bioeng.108:7p1494)。然而,对于药物制剂,一般考虑尽可能减少非活性化合物的使用。这种考虑不但减少产生的药物的成本,而且还减少赋形剂的潜在不需要的作用。例如,因为两性分子的性质和与生物膜反应的能力,许多表面活性剂或多或少是毒性的。通常观察到表面活性剂在水生生物中的LC50低至10mg/L。此外,聚山梨醇酯自氧化或暴露于光可导致形成过氧化氢,其进而可氧化抗体分子,产生不稳定产物(Kerwin,2008;Singh等,2012)。这不仅减少ADC的功效,而且可导致形成ADC的可能有害的降解产物。事实上,作为适合于例如冻干而最初描述于WO2004004639的huC242-DM1ADC的不含表面活性剂的制剂,其包含50mM琥珀酸pH6.0和5.0%蔗糖,后来在WO2007019232A2中报道未充分解决微粒和聚集体形成的问题。因此,对于在运输和贮存期间是稳定的和基本上不含微粒、聚集体和降解产物的ADC,对不含表面活性剂的药物制剂仍有需要。发明简述本发明人已经发现了抗-TFADC的冻干制剂,其中抗-TFADC当重构时保持稳定和不形成聚集体或微粒。非常令人惊讶的是,这些制剂可在不包含表面活性剂例如聚山梨醇酯20或80和/或没有无机盐的情况下制备。因此本发明提供抗-TFADC与药学上可接受的赋形剂的稳定的、不含表面活性剂的冻干制剂,所述赋形剂包含:缓冲液组分,其限制在冻干步骤期间的pH变化;至少一种稳定剂,通常为非还原糖,其与固体状态的抗-TFADC形成非晶相;和至少一种填充剂,任选地其中冻干制剂可基本上不含任何盐。示例性的赋形剂包括但不限于–缓冲液组分,例如组氨酸、柠檬酸盐、琥珀酸盐、乙醇酸盐、碳酸和/或磷酸盐,在冻干之前和/或在重构之后通常提供在水性制剂中约5和约7之间的pH;–一种或多种非还原糖,例如蔗糖和/或海藻糖;–一种或多种填充剂,例如甘露醇和/或甘氨酸。这些和其它方面和实施方案更详细描述于以下部分。附图简述图1显示对于贮存4周的HuMax-TFDOE样品,使用SEC分析得到的百分数HMW(A)、主要(B)和LMW(C)物质。对于这些条柱图,对于各缓冲液加赋形剂亚组,制剂以pH增加的顺序从左到右排列。对于各对条柱,左条柱表示2-8℃和右条柱表示45℃。对于细节,参见实施例2。图2显示对于贮存1周的HuMax-TFDOE样品,各种制剂对使用cIEF观察的酸性(A)、主要(B)和碱性(C)物质的百分数峰面积的影响。对于这些条柱图,对于各缓冲液加赋形剂亚组,制剂以pH增加的顺序从左到右排列。对于各对条柱,左条柱表示2-8℃和右条柱表示45℃。对于细节,参见实施例4。图3显示当在40℃下贮存2周时,通过SEC测定的pH对HuMaxTFADC的百分比HMW的影响。对于细节,参见实施例5。图4显示对在40℃下贮存2周的HuMaxTFADC溶液,通过iCE测定的pH对百分比酸性物质的影响。对于细节,参见实施例5。图5A显示山梨醇和PS80(聚山梨醇酯80)对在pH6.0制备和在40℃贮存2周的HuMaxTFADC溶液的百分比主料峰(通过iCE)的影响。对于细节,参见实施例5。图5B显示山梨醇和PS80(聚山梨醇酯80)对在pH6.0制备和在40℃贮存2周的HuMaxTFADC溶液的主峰(通过SEC)的影响。对于细节,参见实施例5。图6显示液体和冻干制剂之间对在40℃贮存2周的HuMax-TF-ADC的主料同种型百分数的比较。对于细节,参见实施例6。图7显示HuMax-TF-ADC的不同冻干制剂的加速稳定性数据的示例性结果。(A)通过SEC,在50℃时聚集体增加。(B)通过icIEF,在40℃时主料同种型减少。对于细节,参见实施例8。图8显示在40℃下贮存2个月后冻干制剂A、B和C的DLS粒径分布。对于细节,参见实施例8。图9显示在50℃下贮存2周后制剂A、B和C的第二衍生物FTIR谱。数量分布显示不同尺寸组(bin)中的颗粒数。对于细节,参见实施例8。图10显示制剂B的DSC热流温谱图。对于细节,参见实施例11。图11显示用于本发明的ADC制剂的示例性抗-TF抗体的VH和VL序列。根据Kabat的CDR1、CDR2和CDR3序列突出显示:斜体序列表示CDR1区,下划线序列表示CDR2区和粗体序列表示CDR3区。图12显示在40℃下贮存至多2个月后5mg/mL和30mg/mL制剂的SEC平均百分比主峰。参见实施例12。图13显示在40℃下贮存至多2个月后5mg/mL和30mg/mL制剂的SEC平均百分比高分子量物质。参见实施例12。图14显示在40℃下贮存2个月后5mg/mL和30mg/mLHuMax-TF-ADC制剂的iCE百分比主峰。参见实施例12。图15显示在40℃下贮存2个月后5mg/mL和30mg/mLHuMax-TF-ADC制剂的iCE百分比酸性物质。参见实施例12。图16显示在40℃下贮存2个月后5mg/ml和30mg/mlHuMax-TF-ADC制剂的iCE百分比碱性物质。参见实施例12。图17显示对于在25℃下贮存至多48小时的溶液样品,HuMax-TF-ADC的SEC平均%主峰。图18显示对于在25℃下贮存至多48小时的溶液样品,SEC平均%高分子量物质。图19显示对于在25℃下贮存至多48小时的溶液样品,iCE平均%主峰。图20显示对于在25℃下贮存至多48小时的溶液样品,iCE平均%酸性物质。图21显示对于在25℃下贮存至多48小时的溶液样品,iCE平均%碱性物质。图22显示对于在40℃下贮存至多2个月的冻干甘氨酸和柠檬酸盐制剂,SEC平均百分比主峰。图23显示对于在40℃下贮存至多2个月的冻干甘氨酸和柠檬酸盐制剂,SEC平均百分比高分子量物质。图24显示在40℃下贮存2个月后,对于用甘氨酸或柠檬酸盐制备的HuMax-TF-ADC制剂,iCE百分比主峰。图25显示在40℃下贮存2个月后,对于用甘氨酸或柠檬酸盐制备的HuMax-TF-ADC制剂,iCE百分比酸性物质。图26显示在40℃下贮存2个月后,对于用甘氨酸或柠檬酸盐制备的HuMax-TF-ADC制剂,iCE百分比碱性物质。图27显示用pH5或6或7的20mm或50mm组氨酸制备和在40℃下贮存2个月的10mg/mLHuMax-TF-ADC冻干制剂的iCE百分比主峰。图28显示用pH5或6或7的20mm或50mm组氨酸制备和在40℃下贮存2个月的10mg/mLHuMax-TF-ADC制剂的iCE百分比酸性物质。图29显示用pH5或6或7的20mm或50mm组氨酸制备和在40℃下贮存2个月的10mg/mLHuMax-TF-ADC制剂的iCE百分比碱性物质。图30显示用pH5或6或7的20mm或50mm组氨酸制备和在40℃下贮存2个月的10mg/mLHuMax-TF-ADC制剂的SEC百分比主峰。图31显示用pH5或6或7的20mm或50mm组氨酸制备和在40℃下贮存2个月的10mg/mLHuMax-TF-ADC制剂的SEC平均百分比高分子量物质。发明详述定义术语“冻干”和“冷冻干燥”在本文中可互换使用和是指通过首先冷冻和然后减少周围的压力以允许物质中的冷冻的水升华而脱水的物质。本文使用的术语"缓冲液"表示药学上可接受的缓冲液。术语“缓冲液”包括保持溶液的pH值例如在可接受的范围中的那些试剂,和包括但不限于组氨酸、TRIS®(三(羟基甲基)氨基甲烷)、柠檬酸盐、琥珀酸盐、乙醇酸盐等,如本文所述。通常,本文使用的“缓冲液”具有适合于约5-约7、优选地约5.5-6.5的pH范围例如约pH6或约pH6.0的pKa和缓冲能力。术语“填充剂”包括可为冷冻干燥产物提供另外的结构(例如,提供药学上可接受的结块)的试剂。常用的填充剂包括甘露醇、甘氨酸、蔗糖等。除了提供药学上可接受的结块之外,填充剂还通常为冻干组合物提供有用的品质,例如改变塌陷温度(collapsetemperature),提供冻融保护,进一步提高在长期贮存时的蛋白稳定性,等等。这些试剂还可用作张度调节剂。本文使用的术语“稳定剂”包括为蛋白提供稳定性的试剂,例如,用作冷冻期间的冷冻保护剂和/或(冷冻)干燥或“脱水”过程期间的冻干保护剂。合适的稳定剂包括非还原糖或糖类和糖醇例如蔗糖、海藻糖、甘露醇、木糖醇等,以及氨基酸例如甘氨酸、丙氨酸和赖氨酸。稳定剂也可用作填充剂、张度调节剂和/或粘度增加剂。缩写“cIEF”、“icIEF”和“iCE”在本文中可互换使用,全都意指“毛细管等点聚焦”。本文使用的“表面活性剂”是通常用于药物制剂以防止药物吸附至表面和/或聚集的化合物。此外,表面活性剂降低两种液体之间或液体和固体之间的表面张力(或界面张力)。例如,示例性的表面活性剂当以极低浓度(例如,5%w/w或更低、例如3%w/w或更低、例如1%w/w或更低)存在时可显著降低表面张力。表面活性剂是两亲性的,这意思是它们通常由亲水和疏水或亲脂基团两者构成,从而能够在水性溶液中形成胶束或类似的自装配结构。对于药物使用的已知表面活性剂包括甘油单油酸酯、苄索氯铵、多库酯钠、磷脂、聚乙烯烷基醚、月桂基硫酸钠和三辛酸甘油酯(阴离子表面活性剂);苯扎氯铵、十六烷基三甲基溴化铵(citrimide)、西吡氯铵和磷脂(阳离子表面活性剂);和α生育酚、甘油单油酸酯、肉豆蔻醇、磷脂、泊洛沙姆、聚氧乙烯烷基醚、聚氧乙烯蓖麻油衍生物、聚氧乙烯去水山梨糖醇脂肪酸酯、聚氧乙烯硬脂酸酯、聚乙二醇15羟基硬脂酸酯、聚乙二醇甘油酯、聚山梨醇酯、丙二醇二月桂酸酯、丙二醇单月桂酸酯、去水山梨糖醇酯蔗糖棕榈酸酯、蔗糖硬脂酸酯、三辛酸甘油酯和TPGS(非离子和两性离子表面活性剂)。本文的目标“稀释剂”是这样的稀释剂,其为药学上可接受的(对于人类施用是安全和无毒性的)和可用于制备重构制剂。示例性的稀释剂包括无菌水、抑菌注射用水(BWFI)、pH缓冲溶液(例如磷酸盐-缓冲盐水)、无菌盐水溶液、林格溶液或葡萄糖溶液。本文使用的“治疗部分”意指当给予受试者时,特别是当作为本文所述的ADC递送时发挥治疗或预防作用的化合物。“细胞毒性”或“细胞生长抑制”部分是对细胞有害(例如杀死)的化合物。用于ADC的一些细胞毒性或细胞生长抑制部分是疏水的,意味着它们在水中不溶解或仅有有限的溶解度,例如,1g/L或更低(极微溶),例如0.8g/L或更低、例如0.6g/L或更低、例如0.4g/L或更低、例如0.3g/L或更低、例如0.2g/L或更低、例如0.1g/L或更低(几乎不溶)。示例性的疏水性细胞毒性或细胞生长抑制部分包括但不限于某些微管蛋白抑制剂例如奥里斯他汀和其衍生物,例如,MMAF和MMAE。术语“免疫球蛋白”是指一类结构相关的糖蛋白,其由两对多肽链(一对轻(L)链和一对重(H)链)组成,所有四条链通过二硫键互相连接。免疫球蛋白的结构已被充分表征。参见例如FundamentalImmunologyCh.7(Paul,W.等,第二版.RavenPress,N.Y.(1989))。简言之,每条重链通常由重链可变区(本文简称为VH或VH)和重链恒定区(CH或CH)构成。重链恒定区通常由三个结构域CH1、CH2和CH3构成。每条轻链通常由轻链可变区(本文简称为VL或VL)和轻链恒定区(CL或CL)构成。轻链恒定区通常由一个结构域CL构成。VH和VL区可进一步细分为超变性区(或者超变区,其可以在结构限定环的序列和/或形式上是高度变化的),亦称为互补决定区(CDR),其与称为框架区(FR)的更加保守的区交替。各VH和VL通常由三个CDR和四个FR构成,按以下顺序从氨基端至羧基端排列:FR1、CDR1、FR2、CDR2、FR3、CDR3、FR4(亦参见Chothia和LeskJ.Mol.Biol.196,901917(1987))。通常,该区中的氨基酸残基的编号通过Kabat等,SequencesofProteinsofImmunologicalInterest,第5版.PublicHealthService,NationalInstitutesofHealth,Bethesda,MD.(1991)中描述的方法进行(本文的词语例如Kabat中的或根据Kabat的可变结构域残基编号是指用于重链可变结构域或轻链可变结构域的该编号系统)。使用该编号系统,肽的实际线性氨基酸序列可包含对应于可变结构域的FR或CDR的缩短或插入的较少的或另外的氨基酸。例如,重链可变结构域可包括在VHCDR2的残基52后的单个氨基酸插入(按照Kabat的残基52a)和在重链FR残基82后插入的残基(例如按照Kabat的残基82a、82b和82c等)。对于给定的抗体,残基的Kabat编号可通过抗体的序列的同源性区与"标准"Kabat编号序列的比对确定。在本发明的背景中,术语“抗体”(Ab)是指免疫球蛋白分子、免疫球蛋白分子的片段或其任一的衍生物,其具有在典型的生理条件下特异性结合抗原的能力,具有有效的一段时间的半寿期,例如至少约30分钟、至少约45分钟、至少约1小时、至少约2小时、至少约4小时、至少约8小时、至少约12小时、约24小时或更长、约48小时或更长、约3、4、5、6、7或更多天,等等,或任何其它相关功能限定的时间(例如足以诱导、促进、提高和/或调节与抗体和抗原结合有关的生理反应的时间和/或对于抗体足以募集效应器活性的时间)。免疫球蛋白分子的重链和轻链的可变区包含与抗原相互作用的结合结构域。抗体(Ab)的恒定区可介导免疫球蛋白与宿主组织或因子的结合,所述宿主组织或因子包括免疫系统的各种细胞(例如效应细胞)和补体系统的组分例如C1q,补体活化的经典途径的第一组分。如上文所示,除非另外说明或上下文有清楚的相反指示,在本文中术语抗体包括抗体的片段,其保留特异性结合抗原的能力。已经表明抗体的抗原-结合功能可通过全长抗体的片段进行。包括在术语"抗体"内的结合片段的实例包括(i)Fab’或Fab片段,由VL、VH、CL和CH1结构域组成的单价片段,或描述于WO2007059782(GenmabA/S)的单价抗体;(ii)F(ab')2片段,包含在铰链区通过二硫桥连接的两个Fab片段的二价片段;(iii)基本上由VH和CH1结构域组成的Fd片段;(iv)基本上由抗体的单臂的VL和VH结构域组成的Fv片段;(v)dAb片段(Ward等,Nature341,544546(1989)),其基本上由VH结构域组成,亦称为结构域抗体(Holt等;TrendsBiotechnol.2003Nov;21(11):484-90);(vi)骆驼抗体或纳米抗体(Revets等;ExpertOpinBiolTher.2005Jan;5(1):111-24)和(vii)分离的互补决定区(CDR)。此外,尽管Fv片段的两个结构域,VL和VH,通过单独的基因编码,但它们可使用重组方法,通过使得它们能够作为单一蛋白链产生的合成接头而连接,其中VL和VH区配对以形成单价分子(称为单链抗体或单链Fv(scFv),参见例如Bird等,Science242,423426(1988)和Huston等,PNASUSA85,58795883(1988))。这样的单链抗体包括在术语抗体内,除非另外注明或上下文清楚指示。尽管这样的片段通常包括在抗体的含义内,但它们全体和各自独立地是本发明的独特特征,显示不同的生物性质和功用。在本发明的背景中这些和其它有用的抗体片段在本文中进一步论述。还应理解,除非另外说明,术语抗体还包括多克隆抗体、单克隆抗体(mAb)、双特异性抗体、抗体-样多肽,例如嵌合抗体和人源化抗体,和保留特异性结合抗原的能力的抗体片段(抗原-结合片段),其通过任何已知的技术提供,例如酶裂解、肽合成和重组技术。产生的抗体可具有任何同种型。在本发明的背景中,术语“ADC”是指抗体药物缀合物,在本发明的背景中其是指与本申请中描述的另一部分偶联的抗-TF抗体。例如,其可用接头偶联至例如半胱氨酸,或用其它缀合方法偶联至其它氨基酸。所述部分可以是例如药物或毒素等。“抗-TF抗体”是上述的抗体,其特异性结合抗原组织因子或组织因子抗原。术语“组织因子”、“TF”、“CD142”、“组织因子抗原”、“TF抗原”和“CD142抗原”在本文中可互换使用,并且除非另外说明,包括人组织因子的任何变体、同种型和物种同源物,其由细胞天然表达或在用组织因子基因转染的细胞上表达。在一个实施方案中,组织因子氨基酸序列包括Genbank登记号NP_001984.1序列的成熟形式。抗-TF抗体,特别是人抗-TF抗体,可根据描述于WO2011/157741中的方法产生和表征。术语“人抗体”,如本文所用,意欲包括具有源自人种系免疫球蛋白序列的可变区和恒定区的抗体。本发明的人抗体可包括并非由人种系免疫球蛋白序列编码的氨基酸残基(例如,通过体外随机或定点诱变或通过体内体细胞突变引入的突变)。在优选的实施方案中,本发明的ADC的抗体或ADC是分离的。本文使用的"分离的抗体"或“分离的ADC”意欲指基本上不含具有不同的抗原特异性的其它抗体的抗体或ADC(例如,特异性结合TF的分离的抗体基本上不含特异性结合TF以外的抗原的抗体)。本文使用的分离的抗体药物缀合物意欲指这样的抗体药物缀合物,其还基本上不含“游离的毒素”,其中“游离的毒素”意指未与抗体缀合的毒素。具体而言,关于毒素而使用的术语“基本上不含”可意指当按WO2011157741的实施例16所述测定时,存在少于5%、例如少于4%、或少于3%、或少于2%、或少于1.5%、或少于1%、或少于0.5%未缀合的药物。然而,特异性结合人TF的表位、同种型或变体的分离的抗体或分离的抗体药物缀合物可与其它相关抗原,例如来自其它物种的抗原(例如组织因子物种同源物),具有交叉反应。此外,分离的抗体或ADC可基本上不含其它细胞物质和/或化学品。在本发明的一个实施方案中,具有不同的抗原-结合特异性的两种或更多种“分离的”单克隆抗体或ADC在定义明确的组合物中组合。在一个实施方案中,所述两种或更多种分离的单克隆抗体或ADC在两个或更多个不同的表位上结合TF。在另一个实施方案中,其可以是具有对TF的结合特异性的mAb或ADC和具有并非TF的第二结合特异性的一种或多种mAb或ADC的混合物。当本文在两种或更多种抗体的背景下使用时,术语“与……竞争”或“与……交叉竞争”表示抗体与另一抗体,例如“参比”抗体,竞争结合抗原。例如,竞争结合TF的两种或更多种抗体可使用描述于WO10066803的实施例12的测定法进行分析,其中抗体交叉竞争研究使用夹心ELISA进行。简言之,将板各孔用待测试的抗-TF抗体包被过夜(例如在+4℃,使用100微升/孔的含0.5或2微克/ml抗体的PBS缓冲液,用待测试的抗-TF抗体)。ELISA孔用PBS洗涤,在室温下用含2%(v/v)血清(例如,鸡血清)的PBS封闭1小时和用PBS再次洗涤。随后,加入50微升抗-TF参比抗体(10微克/mL),接着加入50微升His-标记的胞外结构域TF(TFECDHis)(0.5或1微克/ml),和在室温下孵育1小时(同时摇振)。将板用PBST(PBS+0.05%tween)洗涤3次,和用1:2000稀释的抗-his-生物素化抗体(例如抗-his生物素BAM050)在室温下孵育1小时(同时摇振)。将板洗涤,和用与直接或间接可检测的化合物缀合的链霉抗生物素(例如,链霉抗生物素-聚-HRP(Sanquin,Amsterdam,TheNetherlands))在室温下孵育20分钟,和再次洗涤。然后,检测和/或定量结合的链霉抗生物素的量。例如,如果间接可检测的化合物是HRP,反应进一步在室温下在暗处用ABTS(RocheDiagnostics)显色,15分钟后通过加入2%(w/v)草酸终止,和测量405nm处的吸光度。该测定也可反转,因为板各孔可用参比抗体包被,然后向其中加入与TF结合的测试抗体。对于一些抗体对,在WO10066803的实施例12的测定法中的竞争仅当一种抗体包被在板上和另一抗体用于竞争时才观察到,反之并非如此。术语“与……竞争”当在本文中使用时还意欲涵盖这样的抗体组合。本文使用的术语"单克隆抗体"或"单克隆抗体组合物"是指具有单一分子组成的抗体分子的制剂。单克隆抗体或其组合物可以是本发明的药物缀合的抗体。单克隆抗体组合物显示对特定表位的单一结合特异性和亲和力。因此,术语"人单克隆抗体"是指显示单一结合特异性的抗体,其具有源自人种系免疫球蛋白序列的可变和恒定区。人单克隆抗体可通过杂交瘤产生,所述杂交瘤包括与永生细胞融合的B细胞,所述B细胞获自具有包含人重链转基因和轻链转基因的基因组的转基因或转染色体的(trans-chromosomal)非人动物,例如转基因小鼠。然后可使用本领域熟知的分子生物学测定人单克隆抗体的cDNA和/或氨基酸序列,使得抗体(任选地具有另一同种型)可重组产生。在抗体与预定抗原结合的背景下,本文使用的术语"结合''或“特异性结合”通常是以对应于以下特定抗体-抗原相互作用的KD(离解平衡常数)的亲和力的结合:当通过例如表面等离子共振(SPR)技术在BIAcore3000仪器上使用抗原作为配体和抗体作为分析物测定时,约10-7M或更低、例如约10-8M或更低、例如约10-9M或更低、约10-10M或更低、或约10-11M或甚至更低,和以对应于以下KD的亲和力结合至预定抗原:比其对结合至并非预定抗原或紧密相关抗原的非特异性抗原(例如,BSA、酪蛋白)的亲和力低至少10倍、例如低至少100倍、例如低至少1,000倍、例如低至少10,000倍、例如低至少100,000倍。亲和力降低的量取决于抗体的KD,使得当抗体的KD很低(即,抗体具有高度特异性)时,则对该抗原的亲和力低于对非特异性抗原的亲和力的量可以是至少10,000倍。在一个实施方案中,本发明还提供包含本文所述的抗体的VL区、VH区或一个或多个CDR的功能变体的抗体的制剂。在抗-TF抗体的背景下使用的VL、VH或CDR的功能变体仍允许抗体保留至少显著比例(至少约50%、60%、70%、80%、90%、95%或更多)的母体抗体的亲和力/亲合力和/或特异性/选择性,和在一些情况下,这样的抗-TF抗体可伴随有比母体抗体更大的亲和力、选择性和/或特异性。这样的功能变体通常保留与母体抗体显著的序列同一性。在两个序列之间的百分比同一性是序列共有的相同位置数的函数(即,%同一性=相同位置数/位置总数x100),考虑了对两个序列的最佳比对需要引入的空位数和各空位的长度。两个序列之间的序列比较和百分比同一性的确定可使用下文描述的数学算法进行。为本发明的目的,在两个氨基酸序列之间的序列同一性使用Needleman-Wunsch算法(Needleman和Wunsch,1970,J.Mol.Biol.48:443-453)确定,在EMBOSS包(EMBOSS:TheEuropeanMolecularBiologyOpenSoftwareSuite,Rice等,2000,TrendsGenet.16:276-277),优选地版本5.0.0或后续版本的Needle程序中执行。使用的参数为:空位开放罚分为10,空位延伸罚分为0.5和EBLOSUM62(BLOSUM62的EMBOSS版本)置换矩阵。Needle标记的"最长同一性"的输出(使用-nobrief选项获得)用作百分比同一性,和如下计算:(相同残基x100)/(比对长度–比对中的空位总数)。为本发明的目的,在两个脱氧核糖核苷酸序列之间的序列同一性使用Needleman-Wunsch算法(Needleman和Wunsch,1970,同上)确定,在EMBOSS包(EMBOSS:TheEuropeanMolecularBiologyOpenSoftwareSuite,Rice等,2000,同上),优选地版本5.0.0或后续版本的Needle程序中执行。使用的参数为:空位开放罚分为10,空位延伸罚分为0.5和EDNAFULL(NCBINUC4.4的EMBOSS版本)置换矩阵。Needle标记的"最长同一性"的输出(使用-nobrief选项获得)用作百分比同一性,和如下计算:(相同脱氧核糖核苷酸x100)/(比对长度–比对中的空位总数)。通过大部分保守取代,CDR变体的序列可不同于母体抗体序列的CDR的序列;例如变体中至少约35%、约50%或更多、约60%或更多、约70%或更多、约75%或更多、约80%或更多、约85%或更多、约90%或更多、约95%或更多、例如约96%、97%或98%或99%的取代是保守氨基酸残基替换。通过大部分保守取代,CDR变体的序列可不同于母体抗体序列的CDR的序列;例如变体中至少10、例如至少9、8、7、6、5、4、3、2或1个取代是保守氨基酸残基替换。在本发明的背景中,保守取代可定义为在以下三个表中的一个或多个所反映的氨基酸类别内的取代:保守取代的氨基酸残基类别备选的保守氨基酸残基取代类别1AST2DE3NQ4RK5ILM6FYW氨基酸残基的备选的物理和功能分类含醇基的残基S和T脂肪族残基I,L,V,和M环烯基相关的残基F,H,W,和Y疏水性残基A,C,F,G,H,I,L,M,R,T,V,W,和Y带负电荷残基D和E极性残基C,D,E,H,K,N,Q,R,S,和T带正电荷残基H,K,和R小残基A,C,D,G,N,P,S,T,和V极小残基A,G,和S参与转角形成的残基A,C,D,E,G,H,K,N,Q,R,S,P,和T柔性残基Q,T,K,S,G,P,D,E,和R较保守的取代分组包括:缬氨酸-亮氨酸-异亮氨酸,苯丙氨酸-酪氨酸,赖氨酸-精氨酸,丙氨酸-缬氨酸,和天冬酰胺-谷氨酰胺。另外的氨基酸组还可使用描述于例如Creighton(1984)Proteins:Structure和MolecularProperties(第2版.1993),W.H.Freeman和Company中的原理进行设计。在本发明的一个实施方案中,与实施例的抗体的CDR相比,关于亲水/吸水性质和残基重量/大小的保守性也基本上保留在变体CDR中(例如,序列的重量类别、亲水评分或两者为至少约50%、至少约60%、至少约70%、至少约75%、至少约80%、至少约85%、至少约90%、至少约95%或更多(例如,约99%)被保留)。例如,保守残基取代可另外或备选地基于强或弱的基于重量的保守基团的替换,这是本领域已知的。类似残基的保留可另外或备选地通过相似性评分来测量,如通过使用BLAST程序所测定的(例如,通过NCBI可得的BLAST2.2.8,使用标准设定BLOSUM62、开放空位=11和延伸空位=1)。合适的变体通常显示与母体肽至少约45%、例如至少约55%、至少约65%、至少约75%、至少约85%、至少约90%、至少约95%或更多(例如,约99%)相似性。本文使用的"同种型"是指由重链恒定区基因编码的免疫球蛋白类别(例如IgG1、IgG2、IgG3、IgG4、IgD、IgA、IgE或IgM)。术语“表位”意指能够特异性结合抗体的蛋白决定簇。表位通常由分子例如氨基酸或糖侧链的表面簇群组成,和通常具有特定的三维结构特征,以及特定的电荷特征。构象和非构象表位的区别在于前者而非后者的结合在变性溶剂的存在下丧失。表位可包含直接参与结合的氨基酸残基(亦称为表位的免疫显性组分)和不直接参与结合的其它氨基酸残基,例如被特异性的抗原结合肽有效阻断的氨基酸残基(换句话说,该氨基酸残基在特异性抗原结合肽的足迹内)。“治疗”是指以减轻、改善、停止或根除(治愈)症状或疾病状态的目的给予有效量的本发明的治疗活性化合物。"有效量"或“治疗有效量”是指在剂量上和必需的时间段内有效实现所需治疗结果的量。抗-TF抗体药物缀合物的治疗有效量可根据因素例如个体的疾病状态、年龄、性别和体重,和抗-TF抗体药物缀合物在个体中引起所需反应的能力而改变。治疗有效量还是其中抗体或抗体部分的任何毒性或有害作用弱于治疗有益作用的量。本发明的具体实施方案本发明至少部分基于以下发现:抗-TFADC的某些水性组合物,当冻干时其提供适用于药物目的和抗-TFADC的治疗应用的稳定的冻干制剂。本文公开的制剂还提供排除表面活性剂例如聚山梨醇酯20和80、无机盐例如NaCl的选项。为了在药物组合物的保质期的时程期间确保功效和安全性,测试该组合物的稳定性。通常,稳定性测试包括但不限于关于组合物的身份、纯度和效力的测试。在预期的贮存温度和在升高的一个或多个温度下测试稳定性。纯度测试可包括但不限于SDS-PAGE、CE-SDS、等电聚焦、免疫电泳、蛋白质印迹、反相色谱、分子排阻色谱(SEC)、离子交换和亲和力色谱。其它测试可包括但不限于:视觉表现,例如颜色和透明度;微粒、pH、水分和重构时间。在稳定性时程期间特别涉及纯度和效力的降解特征,与药物产品的组合物和/或制剂紧密相关。具体而言,制剂的适当选择可显著改变降解特征。表面活性剂例如聚山梨醇酯20经常添加至药物组合物中以降低限制保质期的降解产物的形成。单克隆抗体和源自单克隆抗体的缀合药物产品的典型的降解特征包括共价和非共价高分子量聚集体、片段、脱酰胺和氧化产物的形成。特别是,脱酰胺和氧化产物以及其它酸性物质通常在稳定性测试的时程期间产生。在一些情况下,酸性物质限制药物组合物的可接受的保质期。由于例如脱酰胺导致的酸性物质的形成可通过例如成像毛细管等电聚焦(icIEF)进行测试。在其它情况下,高分子量聚集体的形成限制药物组合物的可接受的保质期。聚集体的形成可通过例如SEC(分子排阻色谱)、DLS、MFI、SDS-PAGE或CE-SDS进行测试。例如,本发明的具有药学上可接受稳定性的抗-TFADC制剂可以是这样的制剂,其中当在约5±3℃或25±2℃的温度下贮存至少约3个月、优选地约6个月和更优选地约12个月或更长、例如18个月或更长、例如至少24或甚至36个月的时间时,当使用SEC分析,例如根据实施例10测定时,聚集体的百分数少于约10%、优选地少于约5%、更优选地少于约2%。另外地或备选地,本发明的稳定的抗-TFADC制剂可以是这样的制剂,其中当在约5±3℃或25±2℃的温度下贮存至少约3个月、优选地约6个月和更优选地约12个月或更长的时间时,当使用icIEF分析,例如根据实施例10测定时,主要的同种型的变化少于15%、优选地少于10%、更优选地少于8%、最优选地少于5%。本发明因此提供以下示例性和非限制性的抗-TFADC的冻干制剂,各自代表具体的实施方案:抗-组织因子(TF)抗体-药物缀合物(ADC)的冻干制剂,所述冻干制剂可通过将包含所述抗-TFADC和药学上可接受的赋形剂的水性制剂冻干获得或通过将包含所述抗-TFADC和药学上可接受的赋形剂的水性制剂冻干获得,其中所述制剂不含表面活性剂。在一个实施方案中药学上可接受的赋形剂包含:a)缓冲液,其限制在冻干步骤期间的pH变化,使得pH保持在5和7之间;b)至少一种非还原糖,其与固体状态的抗-TFADC形成非晶相;和c)至少一种填充剂。在另一个实施方案中,本发明涉及通过冻干包含约5g/L至约30g/L抗-TFADC和药学上可接受的赋形剂的水性制剂制备的冻干制剂,所述赋形剂包含具有约5至约7、任选地在约5.5和约6.5之间的pH的约20至约50mM组氨酸缓冲液;约10至约250mM蔗糖或海藻糖;和约50mM至约300mM甘露醇或甘氨酸。在另一个实施方案中,本发明涉及通过冻干包含约7g/L至约20g/L抗-TFADC和药学上可接受的赋形剂的水性制剂制备的冻干制剂,所述赋形剂包含具有约5至约7、任选地在约5.5和约6.5之间的pH的约20至约50mM组氨酸缓冲液;约10至约250mM蔗糖或海藻糖;和约50mM至约300mM甘露醇或甘氨酸。在另一个实施方案中,本发明涉及通过冻干包含约5g/L至约30g/L抗-TFADC和药学上可接受的赋形剂的水性制剂制备的冻干制剂,所述赋形剂包含具有约5至约7、任选地在约5.5和约6.5之间的pH的约25至约40mM、例如约29至约31mM组氨酸缓冲液;约10至约250mM蔗糖或海藻糖;和约50mM至约300mM甘露醇或甘氨酸。在另一个实施方案中,本发明涉及通过冻干包含约5g/L至约30g/L抗-TFADC和药学上可接受的赋形剂的水性制剂制备的冻干制剂,所述赋形剂包含具有约5至约7、任选地在约5.5和约6.5之间的pH的约20至约50mM组氨酸缓冲液;约50至225mM蔗糖或海藻糖,例如约84至约165mM蔗糖或海藻糖;和约50mM至约300mM甘露醇或甘氨酸。在另一个实施方案中,本发明涉及通过冻干包含约5g/L至约30g/L抗-TFADC和药学上可接受的赋形剂的水性制剂制备的冻干制剂,所述赋形剂包含具有约5至约7、任选地在约5.5和约6.5之间的pH的约20至约50mM组氨酸缓冲液;约10至约250mM蔗糖或海藻糖;和约100mM至约274mM、例如约158至约274、例如约158至约172mM甘露醇或甘氨酸。在另一个实施方案中,本发明涉及通过冻干包含约5g/L至约30g/L抗-TFADC和药学上可接受的赋形剂的水性制剂制备的冻干制剂,所述赋形剂包含具有在约5.5和约6.5之间的pH的约20至约50mM组氨酸缓冲液;约50至约225mM蔗糖或海藻糖;和约100mM至约274mM甘露醇或甘氨酸。在另一个实施方案中,本发明涉及通过冻干包含约5g/L至约30g/L抗-TFADC和药学上可接受的赋形剂的水性制剂制备的冻干制剂,所述赋形剂包含具有约5至约7、任选地在约5.5和约6.5之间的pH的约20至约50mM组氨酸缓冲液;约84至约165mM蔗糖或海藻糖;和约100至约274mM甘露醇或甘氨酸。在另一个实施方案中,本发明涉及通过冻干包含约5g/L至约30g/L抗-TFADC和药学上可接受的赋形剂的水性制剂制备的冻干制剂,所述赋形剂包含具有在约5.5和约6.5之间的pH的约20至约50mM组氨酸缓冲液;约84至约146mM蔗糖;和约158mM至约274mM甘露醇。在另一个实施方案中,本发明涉及通过冻干包含约5g/L至约30g/L抗-TFADC和药学上可接受的赋形剂的水性制剂制备的冻干制剂,所述赋形剂包含具有在约5.5和约6.5之间的pH的约25至约40mM组氨酸缓冲液;约84至约92mM蔗糖;和约158至约274mM甘露醇。在另一个实施方案中,本发明涉及通过冻干包含约7g/L至约20g/L抗-TFADC和药学上可接受的赋形剂的水性制剂制备的冻干制剂,所述赋形剂包含具有在约5.5和约6.5之间的pH的约25至约40mM组氨酸缓冲液;约84至约92mM蔗糖;和约100mM至约274mM甘露醇或甘氨酸。在另一个实施方案中,本发明涉及通过冻干包含约7g/L至约20g/L、例如约9至约11g/L抗-TFADC和药学上可接受的赋形剂的水性制剂制备的冻干制剂,所述赋形剂包含具有约5.5至约6.5的pH的约25至约40mM组氨酸缓冲液;约84至约92mM蔗糖或海藻糖;和约158至约172mM甘露醇或甘氨酸。在另一个实施方案中,本发明涉及通过冻干包含约9至约11g/L抗-TFADC和药学上可接受的赋形剂的水性制剂制备的冻干制剂,所述赋形剂包含具有约5.5至约6.5的pH的约29至约31mM组氨酸缓冲液;约84至约92mM蔗糖;和约158至约172mM甘露醇或甘氨酸。在另一个实施方案中,本发明涉及通过冻干包含约9至约11g/L抗-TFADC、例如约10mg/mL抗-TFADC和药学上可接受的赋形剂的水性制剂制备的冻干制剂,所述赋形剂包含具有约6的pH的约30mM组氨酸缓冲液;约88mM蔗糖;和约165mM甘露醇或甘氨酸。在另一个实施方案中,本发明涉及通过冻干包含约10mg/mL抗-TFADC和药学上可接受的赋形剂的水性制剂制备的冻干制剂,所述赋形剂包含约25至约40mM组氨酸缓冲液、例如30-35mM组氨酸缓冲液、例如约30mM,和具有约6的pH;约88mM蔗糖;和约165mM甘露醇。在另一个实施方案中,本发明涉及通过冻干水性制剂制备的冻干制剂,所述水性制剂包含约9至约11g/L抗-TFADC、例如约10mg/mL抗-TFADC,其中抗-TF-ADC是HuMaxTFADC(IgG1,vcMMAE),其是由通过蛋白酶可裂解的缬氨酸瓜氨酸接头与药物单甲基奥里斯他汀E(vcMMAE)缀合的针对TF的人单克隆IgG1,κ抗体011构成的ADC;和药学上可接受的赋形剂,其包含具有约6的pH的约30mM组氨酸缓冲液;约88mM蔗糖;和约165mM甘露醇或甘氨酸。在单独和特定的实施方案中,该制剂基本上不含表面活性剂。在本发明的又一方面中,该制剂不含表面活性剂。在另一个实施方案中,本发明涉及抗-TFADC的冻干制剂,所述冻干制剂通过冻干水性制剂制备,所述水性制剂包含药学上可接受的赋形剂,所述赋形剂包含:缓冲液,其限制在冻干步骤期间的pH变化;至少一种非还原糖,其与固体状态的抗-TFADC形成非晶相;和至少一种填充剂,其中抗-TFADC包含药物-接头,其选自mcMMAF、mcMMAE、vcMMAF和vcMMAE;和抗-TF抗体,其包含VH和VL区,选自:包含SEQIDNO:5的氨基酸序列的VH区和包含SEQIDNO:45的氨基酸序列的VL区;包含SEQIDNO:33的氨基酸序列的VH区和包含SEQIDNO:73的氨基酸序列的VL区;包含SEQIDNO:37的氨基酸序列的VH区和包含SEQIDNO:77的氨基酸序列的VL区;或包含SEQIDNO:1的氨基酸序列的VH区和包含SEQIDNO:41的氨基酸序列的VL区;任选地其中冻干制剂基本上不包含任何表面活性剂。在单独和特定的实施方案中,前述实施方案中任一个的ADC的抗体-部分包含人抗-TF抗体011的VH和VLCDR,任选地VH(SEQIDNO:5)和VL(SEQIDNO:45)序列,和药物-接头,其为mcMMAE、vcMMAE、vcMMAF或mcMMAF。在单独和特定的实施方案中,前述实施方案中任一个的ADC的抗体-部分包含人抗-TF抗体098的VH和VLCDR,任选地VH(SEQIDNO:33)和VL(SEQIDNO:73)序列,和药物-接头,其为mcMMAE、vcMMAE、vcMMAF或mcMMAF。在单独和特定的实施方案中,前述实施方案中任一个的ADC的抗体-部分包含人抗-TF抗体111的VH和VLCDR,任选地VH(SEQIDNO:37)和VL(SEQIDNO:77)序列,和药物-接头,其为mcMMAE、vcMMAE、vcMMAF或mcMMAF。在其它单独和特定的实施方案中,本发明提供前述实施方案中任一个的冻干制剂,其基本上不含任何聚山梨醇酯,任选地不含任何表面活性剂。本发明还提供基本上由以下组成的冻干制剂:抗-TF抗体药物缀合物;选自组氨酸、柠檬酸盐和tris的缓冲剂;选自蔗糖、海藻糖和其组合的非还原糖;和选自甘露醇和甘氨酸的填充剂。在一个实施方案中,抗-TF抗体包含具有选自以下的VH和VL序列的VH和VLCDR区,任选地VH和VL序列:包含SEQIDNO:5的氨基酸序列的VH区和包含SEQIDNO:45的氨基酸序列的VL区;包含SEQIDNO:33的氨基酸序列的VH区和包含SEQIDNO:73的氨基酸序列的VL区;包含SEQIDNO:37的氨基酸序列的VH区和包含SEQIDNO:77的氨基酸序列的VL区;或包含SEQIDNO:1的氨基酸序列的VH区和包含SEQIDNO:41的氨基酸序列的VL区。在一个实施方案中,抗-TFADC分别包含SEQIDNOS:5和45的VH和VLCDR。在一个实施方案中,抗-TFADC分别包含SEQIDNOS:33和73的VH和VLCDR。在一个实施方案中,抗-TFADC分别包含SEQIDNOS:37和77的VH和VLCDR。在前述实施方案中任一个的一个特定的实施方案中,前述实施方案中任一个的冻干制剂包含甘露醇和蔗糖,其中甘露醇与蔗糖的重量比是至少约1,例如在约1和约30之间,例如在1和约10之间,例如在约1和约2之间,例如约1。在前述实施方案中任一个的一个特定的实施方案中,前述实施方案中任一个的冻干制剂包含甘露醇和海藻糖,其中甘露醇与海藻糖的重量比是至少约1,例如在约1和约30之间,例如在1和约10之间,例如在约1和约2之间,例如约1。在前述实施方案中任一个的一个特定的实施方案中,前述实施方案中任一个的冻干制剂包含甘露醇和蔗糖,其中甘露醇与蔗糖的重量比是在约1和约10之间和甘露醇与ADC的重量比是至少约3,例如在3和30之间。本发明还提供通过冻干水性制剂可获得的冻干制剂,所述水性制剂包含(任选地由其组成)约9至约11g/L抗-TFADC和约30mM组氨酸;约88mM蔗糖;和约165mM甘露醇。本发明还提供适合于制备抗-TFADC的冻干制剂的水性溶液,包含a.约7至约20g/L抗-TFADC,任选包含抗-TF抗体011的VH和VLCDR或VH和VL序列;b.约28至34mM组氨酸;c.约84至约146mM蔗糖;d.约158至约274mM甘露醇;或e.a)和(b)至(d)中的任何两个、三个或全部的组合。本发明还提供适合于制备抗-TFADC的冻干制剂的水性溶液,其中所述水性溶液不包含表面活性剂,所述溶液包含:a.约7至约20g/L抗-TFADC,任选地包含抗-TF抗体011的VH和VLCDR或VH和VL序列;b.约28至34mM组氨酸;c.约84至约146mM蔗糖;d.约158至约274mM甘露醇;或e.a)和(b)至(d)中的任何两个、三个或全部的组合。本发明还提供适合于制备抗-TFADC的冻干制剂的水性溶液,其由以下组成:a.约7至约20g/L抗-TFADC,任选地包含抗-TF抗体011的VH和VLCDR或VH和VL序列;b.约28至34mM组氨酸;c.约84至约146mM蔗糖;d.约158至约274mM甘露醇;或e.a)和(b)至(d)中的任何两个、三个或全部的组合。本发明还提供通过在无菌水性稀释剂中重构前述方面或实施方案中任一个的冻干制剂获得的药学上可接受的液体制剂。例如,这样的液体制剂可包含约5g/L至约30g/L抗-TFADC、具有约5至约7的pH的约20至约50mM组氨酸;约10至约250mM蔗糖或海藻糖;和约50mM至约300mM甘露醇或甘氨酸,或基本上由其组成。在一个实施方案中,液体制剂包含约9至约11mg/mL抗-TFADC、约28至约34mM组氨酸、约84至约92mM蔗糖和约158至约274mM甘露醇,或基本上由其组成。抗-TFADC的冻干制剂可通过冻干包含约9g/L至约11g/L抗-TFADC和药学上可接受的赋形剂的水性制剂制备,所述赋形剂包含:具有约5.5至约6.5的pH的约30mM组氨酸缓冲液;约88mM蔗糖;和约165mM甘露醇;其中抗体包含:包含SEQIDNO:5的氨基酸序列的VH区和包含SEQIDNO:45的氨基酸序列的VL区;包含SEQIDNO:33的氨基酸序列的VH区和包含SEQIDNO:73的氨基酸序列的VL区;包含SEQIDNO:37的氨基酸序列的VH区和包含SEQIDNO:77的氨基酸序列的VL区;或包含SEQIDNO:1的氨基酸序列的VH区和包含SEQIDNO:41的氨基酸序列的VL区,和其中药物是MMAF或MMAE,例如,为vcMMAE的接头-药物。在另一个实施方案中,本发明的冻干制剂包含少于3.0wt.%水分。在另一个实施方案中,本发明的冻干制剂包含少于2.0wt.%水分。在另一个实施方案中,本发明的冻干制剂包含少于1.0wt.%水分。在另一个实施方案中,本发明的冻干制剂包含少于0.5wt.%水分。在另一个优选的方面,上述任一种优选的制剂包含确切数量的其中所包含的一种或多种组分和/或确切的pH值。换句话说,在本发明的该其它优选方面中,一个或多个术语"约"被删除。抗体药物-缀合物如本文所述,本发明的制剂适合于例如抗-TFADC。在一个实施方案中,本发明的冻干制剂包含与治疗部分缀合的抗-TFADC,所述治疗部分选自泰素;细胞松弛素B;短杆菌肽D;溴化乙锭;吐根素;丝裂霉素;依托泊苷;替尼泊苷(tenoposide);长春新碱;长春碱;秋水仙碱;多柔比星;柔红霉素;二羟基炭疽菌素二酮;微管蛋白抑制剂例如美登素或其类似物或衍生物;米托蒽醌;普卡霉素;放线菌素D;1–去氢睾酮;糖皮质激素;普鲁卡因;丁卡因;利多卡因;普萘洛尔;嘌罗霉素;刺孢霉素或其类似物或衍生物;抗代谢物例如甲氨蝶呤、6巯嘌呤、6硫鸟嘌呤、阿糖胞苷、氟达拉滨、5氟尿嘧啶、氨烯咪胺、羟基脲、门冬酰胺酶、吉西他滨或克拉屈滨;烷化剂例如氮芥、thioepa、苯丁酸氮芥、美法仑、卡莫司汀(BSNU)、洛莫司汀(CCNU)、环磷酰胺、白消安、二溴甘露醇、链唑霉素、达卡巴嗪(DTIC)、丙卡巴肼、丝裂霉素C、顺铂、卡铂、倍癌霉素(duocarmycin)A、倍癌霉素SA、rachelmycin(CC-1065)或其类似物或衍生物;吡咯并[2,1-c][1,4]苯并二氮杂(PDBs)或其类似物;抗生素例如更生霉素、博来霉素、柔红霉素、多柔比星、伊达比星、光神霉素、丝裂霉素、米托蒽醌、普卡霉素、安曲霉素(AMC));白喉毒素和相关分子例如白喉A链和其活性片段和杂合分子、蓖麻毒素例如蓖麻毒素A或去糖基化蓖麻毒素A链毒素、霍乱毒素、志贺-样毒素例如SLTI、SLTII、SLTIIV、LT毒素、C3毒素、志贺毒素、百日咳毒素、破伤风毒素、大豆Bowman-Birk蛋白酶抑制剂、假单胞菌外毒素、芦荟素(alorin)、皂草素、蒴莲根毒素、gelanin、相思豆毒素A链、蒴莲根毒素A链、α-八叠球菌素、油桐蛋白、石竹素蛋白、美洲商陆蛋白例如PAPI、PAPII和PAPS、苦瓜蛋白抑制剂、麻疯树毒蛋白、巴豆毒蛋白、芦荀(sapaonariaofficinalis)蛋白抑制剂、白树毒素、mitogellin、局限曲菌素、酚霉素和依诺霉素毒素;核糖核酸酶(RNase);DNaseI、葡萄球菌肠毒素A;商陆抗病毒蛋白;白喉毒素;和假单胞菌内毒素。在一个实施方案中,抗体与细胞毒性或细胞生长抑制部分缀合,所述部分为选自多拉司他汀、美登素、刺孢霉素、倍癌霉素、rachelmycin(CC-1065)或其任一的类似物、衍生物或前药的药物。在一个实施方案中,抗体与治疗性、细胞生长抑制和/或细胞毒性的部分缀合,所述部分为微管蛋白抑制剂、DNA相互作用化合物和/或激酶抑制剂。在一个实施方案中,抗体与疏水性化合物缀合,例如疏水性微管蛋白抑制剂,优选地奥里斯他汀,更优选MMAE或MMAF,和最优选MMAE。药物-载量(或,每抗体分子的细胞生长抑制或细胞毒性药物的平均数)通常为1-约8,例如p可以是3-6,例如4-6或3-5,或p可以是1、2、3、4、5、6、7或8,例如3、4或5,例如4。用于本发明的制剂的ADC通常在细胞生长抑制或细胞毒性药物单元和抗体单元之间包含接头单元。在一些实施方案中,接头可在细胞内条件下裂解,使得在细胞内环境中接头的裂解从抗体释放药物单元。在又一实施方案中,接头单元不可裂解,和药物通过例如抗体降解而释放。在一些实施方案中,接头可通过在细胞内环境中(例如在溶酶体或内体或胞膜窖内)存在的裂解剂而裂解。接头可以是例如肽基接头,其通过细胞内的肽酶或蛋白酶裂解,包括但不限于溶酶体或内体蛋白酶。在一些实施方案中,肽基接头为至少2个氨基酸长或至少3个氨基酸长。裂解剂可包括组织蛋白酶B和D和纤溶酶,已知所有这些水解二肽药物衍生物,导致在靶细胞内释放活性药物(参见例如Dubowchik和Walker,1999,Pharm.Therapeutics83:67-123)。在特定的实施方案中,可通过细胞内蛋白酶裂解的肽基接头是Val-Cit(缬氨酸-瓜氨酸)接头或Phe-Lys(苯丙氨酸-赖氨酸)接头(参见例如US6214345,其描述了具有Val-Cit接头的多柔比星的合成和Phe-Lys接头的不同实例)。Val-Cit和Phe-Lys接头的结构的实例包括但不限于下文描述的MC-vc-PAB、MC-vc-GABA、MC-Phe-Lys-PAB或MC-Phe-Lys-GABA,其中MC是马来酰亚胺基己酰基的缩写,vc是Val-Cit的缩写,PAB是对氨基苄基氨基甲酸酯的缩写,和GABA是γ-氨基丁酸的缩写。使用治疗剂的细胞内蛋白水解释放的优点是当缀合时药剂通常是减毒的,和缀合物的血清稳定性通常高。在一些实施方案中,接头单元不可裂解,和药物通过抗体降解释放(参见US2005/0238649)。通常,这样的接头不对细胞外环境显著敏感。在优选的实施方案中,抗体与多拉司他汀衍生物例如奥里斯他汀缀合。奥里斯他汀或奥里斯他汀肽类似物和衍生物已经显示干扰微管动力学、GTP水解和核和细胞分裂,并具有抗-癌和抗-真菌活性,描述于例如US5635483;US5780588;US5663149;其所有通过引用以其整体结合到本文中。奥里斯他汀药物部分通常与抗体经过接头,通过肽药物部分的N(氨基)端或C(端)连接。示例性的奥里斯他汀实施方案包括N-端-连接的单甲基奥里斯他汀药物部分DE和DF,公开于Senter等,ProceedingsoftheAmericanAssociationforCancerResearch.第45卷,摘要号623,2004年3月28日提供和描述于US2005/0238649)。在一个实施方案中奥里斯他汀是单甲基奥里斯他汀E(MMAE):其中波浪线表示接头的连接位点。在一个实施方案中奥里斯他汀是单甲基奥里斯他汀F(MMAF):其中波浪线表示接头的连接位点。在一个实施方案中接头与抗体例如抗-TF抗体的巯基残基连接,其通过抗体的(部分)还原获得。在一个实施方案中接头-奥里斯他汀是MC-vc-PAB-MMAF(亦称为vcMMAF)或MC-vc-PAB-MMAE(亦称为vcMMAE):Ab-MC-vc-PAB-MMAF(Ab-vcMMAF)Ab-MC-vc-PAB-MMAE(Ab-vcMMAE)其中p表示1-8的数字,例如p可以是3-5,S表示抗体的巯基残基,和Ab表示抗体。在一个实施方案中接头-奥里斯他汀是vcMMAE。在一个实施方案中接头-缀合物是mcMMAF(其中mc/MC是马来酰亚胺基己酰基的缩写):Ab-MC-MMAF(Ab-mcMMAF)其中p表示1-8的数字,例如p可以是3-5,S表示抗-TF抗体的巯基残基,和Ab表示抗体。尽管一般可适用于任何抗-TF抗体,但特别适用于本发明的ADC制剂的抗体是与本文提供了其VH和VL序列(参见表1和图11)的任何一种或多种抗-TF抗体,例如抗体011、098、114、017-D12、042、092-A09、101、025、109或111,例如抗体011、098或111,例如011,共有一个或多个物理化学和/或抗原-结合性质的那些抗体。因此,在一个实施方案中,当与所述药物缀合时,得到的ADC可具有范围为约5至约12、例如约7至约10、例如约8.5至约9.5、例如约8.5至约9.0的pI。在一个实施方案中,ADC可结合与人抗体011相同的表位,和/或包含人抗体011的一个或多个CDR序列,所述抗体任选地呈IgG1,κ形式。在一个实施方案中,ADC可结合与人抗体098相同的表位,和/或包含人抗体098的一个或多个CDR序列,所述抗体任选地呈IgG1,κ形式。在一个实施方案中,ADC可结合与人抗体114相同的表位,和/或包含人抗体114的一个或多个CDR序列,所述抗体任选地呈IgG1,κ形式。在一个实施方案中,ADC可结合与人抗体017-D12相同的表位,和/或包含人抗体017-D12的一个或多个CDR序列,所述抗体任选地呈IgG1,κ形式。在一个实施方案中,ADC可结合与人抗体042相同的表位,和/或包含人抗体042的一个或多个CDR序列,所述抗体任选地呈IgG1,κ形式。在一个实施方案中,ADC可结合与人抗体092-A09相同的表位,和/或包含人抗体092-A09的一个或多个CDR序列,所述抗体任选地呈IgG1,κ形式。在一个实施方案中,ADC可结合与人抗体101相同的表位,和/或包含人抗体101的一个或多个CDR序列,所述抗体任选地呈IgG1,κ形式。在一个实施方案中,ADC可结合与人抗体025相同的表位,和/或包含人抗体025的一个或多个CDR序列,所述抗体任选地呈IgG1,κ形式。在一个实施方案中,ADC可结合与人抗体109相同的表位,和/或包含人抗体109的一个或多个CDR序列,所述抗体任选地呈IgG1,κ形式。在一个实施方案中,ADC可结合与人抗体111相同的表位,和/或包含人抗体111的一个或多个CDR序列,所述抗体任选地呈IgG1,κ形式。在一个实施方案中,抗体是与包含包含SEQIDNO:33的序列的VH区和包含SEQIDNO:73的序列的VL区的参比抗体,或与包含包含SEQIDNO:1序列的VH区和包含SEQIDNO:41序列的VL区的抗体,或与包含包含SEQIDNO:5的序列的VH区和包含SEQIDNO:45的序列的VL区的抗体,或与包含包含SEQIDNO:9的序列的VH区和包含SEQIDNO:49的序列的VL区的抗体,或与包含包含SEQIDNO:13的序列的VH区和包含SEQIDNO:53的序列的VL区的抗体,或与包含包含SEQIDNO:17的序列的VH区和包含SEQIDNO:57的序列的VL区的抗体,或与包含包含SEQIDNO:21的序列的VH区和包含SEQIDNO:61的序列的VL区的抗体,或与包含包含SEQIDNO:25的序列的VH区和包含SEQIDNO:65的序列的VL区的抗体,或与包含包含SEQIDNO:29的序列的VH区和包含SEQIDNO:69的序列的VL区的抗体,或与包含包含SEQIDNO:37的序列的VH区和包含SEQIDNO:77的序列的VL区的抗体,竞争组织因子结合的抗-TF抗体。在一个实施方案中,抗体是与包含包含SEQIDNO:5的序列的VH区和包含SEQIDNO:45的序列的VL区的参比抗体竞争组织因子结合的抗-TF抗体。在一个实施方案中,抗-TF抗体包含:包含SEQIDNO:34、35和36的CDR1、2和3序列的VH区和包含SEQIDNO:74、75和76的CDR1、2和3序列的VL区;或包含SEQIDNO:2、3和4的CDR1、2和3序列的VH区和包含SEQIDNO:42、43和44的CDR1、2和3序列的VL区;或包含SEQIDNO:6、7和8的CDR1、2和3序列的VH区和包含SEQIDNO:46、47和48的CDR1、2和3序列的VL区;或包含SEQIDNO:10、11和12的CDR1、2和3序列的VH区和包含SEQIDNO:50、51和52的CDR1、2和3序列的VL区;或包含SEQIDNO:14、15和16的CDR1、2和3序列的VH区和包含SEQIDNO:54、55和56的CDR1、2和3序列的VL区;或包含SEQIDNO:18、19和20的CDR1、2和3序列的VH区和包含SEQIDNO:58、59和60的CDR1、2和3序列的VL区;或包含SEQIDNO:22、23和24的CDR1、2和3序列的VH区和包含SEQIDNO:62、63和64的CDR1、2和3序列的VL区;或包含SEQIDNO:26、27和28的CDR1、2和3序列的VH区和包含SEQIDNO:66、67和68的CDR1、2和3序列的VL区;或包含SEQIDNO:30、31和32的CDR1、2和3序列的VH区和包含SEQIDNO:70、71和72的CDR1、2和3序列的VL区;或包含SEQIDNO:38、39和40的CDR1、2和3序列的VH区和包含SEQIDNO:78、79和80的CDR1、2和3序列的VL区;或任何所述抗体的变体,其中所述变体优选地在所述序列中具有至多1、2或3个氨基酸修饰,更优选地氨基酸取代,例如保守氨基酸取代。在一个实施方案中,抗-TF抗体包含:包含SEQIDNO:6、7和8的CDR1、2和3序列的VH区和包含SEQIDNO:46、47和48的CDR1、2和3序列的VL区,或在所述序列中具有至多1、2或3个氨基酸修饰,更优选地氨基酸取代,例如保守氨基酸取代的变体。在一个实施方案中,抗-TF抗体包含与选自以下的VH区序列具有至少80%同一性、例如至少90%、至少95%、或至少98%或100%同一性的VH:SEQIDNO:33、1、5、9、13、17、21、25、37和29;或与选自以下的VH区序列相比,具有至多20、例如15、或10、或5、4、3、2或1个氨基酸修饰,更优选地氨基酸取代,例如保守氨基酸取代:SEQIDNO:1、5、9、13、17、21、25、33、37和29。在一个实施方案中,抗-TF抗体包含与选自以下的VL区序列具有至少80%同一性、例如至少90%、至少95%、或至少98%或100%同一性的VL:SEQIDNO:41、45、49、53、57、61、65、73、77和69;或与选自以下的VH区序列相比,具有至多20、例如15、或10、或5、4、3、2或1个氨基酸修饰,更优选地氨基酸取代,例如保守氨基酸取代:SEQIDNO:41、45、49、53、57、61、65、73、77和69。在单独和特定的实施方案中,抗-TF抗体是全长的完全人单克隆IgG1,κ抗体抗-TFHuMab092-A09、抗-TFHuMab101、抗-TFHuMab025、抗-TFHuMab109、抗-TFHuMab017-D12、抗-TFHuMab114、抗-TFHuMab042、抗-TFHuMab011、抗-TFHuMab098或抗-TFHuMab111或包含其任一的VH和VLCDR的抗体,或包含其VH和VL序列的抗体。在一个具体的实施方案中,抗-TF抗体是抗-TFHuMab011,或包含其VHCDR1、2、3和VLCDR1、2、3序列的抗体,或包含其VH和VL序列的抗体。在一个具体的实施方案中,抗-TF抗体是抗-TFHuMab098,或包含其VHCDR1、2、3和VLCDR1、2、3序列的抗体,或包含其VH和VL序列的抗体。在一个具体的实施方案中,抗-TF抗体是抗-TFHuMab111,或包含其VHCDR1、2、3和VLCDR1、2、3序列的抗体,或包含其VH和VL序列的抗体。表1显示关于这些抗体的VH(1A)和VL(1B)序列的具体的序列标识符(SEQIDNo)。在特别优选的实施方案中,ADC是HuMaxTFADC(IgG1,vcMMAE),其为由通过蛋白酶可裂解的缬氨酸瓜氨酸接头与药物单甲基奥里斯他汀E(vcMMAE)缀合的针对TF的人单克隆IgG1,κ抗体011构成的ADC。该抗体的鉴定和生产描述于WO2011157741。各单克隆抗体(mAb)分子带有平均4个药物分子。抗体部分具有147kDa的近似分子量。平均4个分子的vcMMAE(分子量1.3kDa)连接至各mAb分子,得到HuMaxTFADC的总平均分子量为152kDa。HuMax-TF-ADC的等电点为大约8.7。制剂根据本发明使用的抗体的治疗制剂通过混合具有需要的纯度的ADC与任选的药学上可接受的载体、赋形剂或稳定剂以冻干制剂或水性溶液的形式制备用于贮存(Remington'sPharmaceuticalSciences第16版,Osol,A.编辑(1980))。可接受的载体、赋形剂或稳定剂以所用的剂量和浓度对接受者是无毒的。一般而言,本发明的冻干和重构制剂包含抗-TFADC、缓冲剂、稳定剂(通常为非还原糖或糖醇或氨基酸)和填充剂。优选的稳定剂是蔗糖、海藻糖和其组合。优选的填充剂是甘露醇、甘氨酸和其组合。本文使用的术语"缓冲液"表示药学上可接受的缓冲液。一般而言,缓冲液具有适合于约5-约7的pH范围的pKa和缓冲能力,优选地约5.5-6.5,例如约pH6或约pH6.0。对于冻干制剂,缓冲液组分不应在亚环境温度下在使用的浓度下结晶。具有较高塌陷温度的缓冲液是优选的,因为其将使得更快和更稳健的冻干循环成为可能。合适的药学上可接受的缓冲液包括但不限于组氨酸-缓冲液、柠檬酸盐-缓冲液、琥珀酸盐-缓冲液、碳酸-缓冲液、磷酸盐缓冲液、乙醇酸盐-缓冲液、TRIS®(三(羟基甲基)氨基甲烷)缓冲液和其混合物。优选的缓冲液基于L-组氨酸、柠檬酸盐、磷酸盐、碳酸、琥珀酸盐和/或乙醇酸盐,例如组氨酸和/或柠檬酸盐,和还包括混合物,例如,L-组氨酸与L-组氨酸盐酸盐或与TRIS®(三(羟基甲基)氨基甲烷)的混合物。可能的是,可能需要用本领域已知的酸或碱调整pH。上述L-组氨酸、柠檬酸盐、磷酸盐、碳酸、琥珀酸盐和/或乙醇酸盐缓冲液通常以约1mM至约100mM、例如约5至约80mM、优选地约20mM至约50mM、更优选地约10至约30mM和还更优选地约30mM的量使用。磷酸盐缓冲液的浓度优选地在约1至约30mM的范围内。不依赖于使用的缓冲液,通过用本领域已知的酸或碱调整或通过使用缓冲液组分的适当混合物或两者,pH可调整在包括约5至约7的值,和优选地约5.5至约6.5和最优选地约6.0。优选地,缓冲液包含浓度为约10至约30mM的组氨酸和/或柠檬酸盐缓冲液,例如浓度为约30mM的组氨酸缓冲液。本发明的制剂可进一步包含上文定义的一种或多种药学上可接受的稳定剂和本领域亦称为"冻干保护剂"的成分,例如糖、糖醇、氨基糖、氨基酸和葡聚糖,如本领域已知的。通常,药学上可接受的稳定剂可以约1mM至约500mM的量使用。合适的糖包括但不限于单糖和二糖。根据本发明使用的糖和糖醇的非限制性实例包括海藻糖、蔗糖、甘露醇、山梨醇、甘露糖、麦芽糖、半乳糖、果糖、山梨糖、棉子糖、葡糖胺、N-甲基葡糖胺(亦称为"葡甲胺")、半乳糖胺和神经氨酸和其组合。优选的是非还原糖和糖醇,例如蔗糖或海藻糖,浓度为约10至约250mM,例如约50至225mM、例如约84至146、例如约84至92mM。最优选蔗糖。在一个实施方案中,制剂包含84至92mM蔗糖,例如85、86、87、88、89、90、91或92mM蔗糖。最优选制剂包含88mM蔗糖。特别是,糖醇例如甘露醇还可用作填充剂以产生均质和稳定的冻干结块,其可在可接受的时间内重构,更具体在0–600秒内。通常,“填充剂”在当API的总量太小而不能为结块提供适当的结构时使用。填充剂应提供惰性基质,其产生药学上精致的结块。填充剂还改变制剂的热学性质。活性药物的浓度经常太低,以致系统的冷冻-干燥性质仅归因于填充剂。常用的填充剂包括甘露醇、甘氨酸作为结晶填充剂;蔗糖、海藻糖、明胶、葡聚糖作为非结晶填充剂。优选的填充剂是甘露醇和甘氨酸。在一个实施方案中,制剂包含约158至约274mM甘露醇,例如160mM或162mM或165mM或170mM或180mM或200mM甘露醇。最优选其包含约165mM甘露醇或165mM甘露醇。本发明的某些冻干制剂经设计使得有可能排除表面活性剂。然而,本领域技术人员可理解,对于一些目的可能仍然期需包括表面活性剂。合适的药学上可接受的表面活性剂包括但不限于聚乙烯-去水山梨糖醇-脂肪酸酯、聚乙烯-聚丙烯二醇、聚氧乙烯-硬脂酸酯和十二烷基硫酸钠。聚乙烯-去水山梨糖醇-脂肪酸酯包括聚乙烯(20)-去水山梨糖醇-酯(与聚山梨醇酯20同义,以商品名(TM)Tween20(TM)出售),和聚氧乙烯(20)去水山梨糖醇单油酸酯(与聚山梨醇酯80同义,以商品名Tween80(TM)出售)。聚乙烯-聚丙烯二醇为以名称Pluronic(R)F68或Poloxamer188(TM)出售的那些。聚氧乙烯-硬脂酸酯以商品名Myrj(TM)出售。聚氧乙烯单月桂基醚为以商品名Brij(TM)出售的那些。需要时,可使用聚乙烯-去水山梨糖醇-聚乙烯(20)-去水山梨糖醇-酯(Tween20(TM))和聚氧乙烯(20)去水山梨糖醇单油酸酯(Tween80(TM)),例如,其量为约0.01%至约0.06%,例如约0.02%至约0.04%。本发明的某些冻干制剂经设计,使得有可能从冻干前液体和冻干制剂中排除无机盐,例如氯化钠(NaCl),其经常用作等渗剂。盐的其它实例包括阳离子钠、钾、钙或镁与阴离子氯离子、磷酸根、柠檬酸根、琥珀酸根、硫酸根或其混合物的任何组合的盐。然而,本领域技术人员能理解,对于一些目的,可能仍期需包括无机盐,例如,对于冻干制剂的重构,即作为下文描述的稀释剂。本发明的制剂可还包含一种或多种以下成分:抗氧化剂、抗坏血酸、谷胱甘肽、防腐剂(例如十八烷基二甲基苄基氯化铵;氯己双铵;苯扎氯铵;苄索氯铵;苯酚、丁醇或苯甲醇;对羟基苯甲酸烷基酯例如对羟基苯甲酸甲酯或丙酯;儿茶酚;间苯二酚;环己醇;3-戊醇;和间甲酚);环糊精,例如羟基丙基--环糊精、磺基丁基乙基--环糊精、[β]-环糊精;聚乙二醇例如PEG3000、3350、4000或6000;低分子量(少于约10个残基)多肽;蛋白,例如血清白蛋白、明胶或免疫球蛋白;螯合剂例如EDTA;成盐抗衡离子例如钠;和金属复合物(例如Zn-蛋白复合物)。方法冷冻干燥通常包括三个主要步骤:冷冻、初级干燥和次级干燥。第一阶段用于产物在低于产物的共晶或玻璃转变温度的温度下冷冻。冷冻速率影响水结晶的大小和随后的干燥速率。第二阶段是初级干燥,其除去溶剂(冰)水。重要的是,产物温度保持低于塌陷温度和所有的冰/水被升华。第三阶段是次级干燥,其除去“结合的”水或来自溶质的水,在此期间贮存温度经常升高高于40℃以加速解吸过程。适用于抗体和其它蛋白或蛋白缀合物制剂的冻干方法是本领域技术人员熟知的,和描述于例如HenryR.Costantino和MichaelJ.Pikal的“LyophilizationofBiopharmaceuticals”;LouisRey和JoanC.May的“FreezeDrying/LyophilizationofPharmaceuticalsandBiologicalProducts”。在本发明的一个方面中,水性溶液的冻干包括以下步骤:a.将水性溶液以0.3℃/min至3℃/min冷却至-40℃和-60之间;b.等温保持至少120min;c.以0.3℃/min至6℃/min的速率升温至-20℃和-15℃之间;d.等温保持至少180min;e.使用30mTorr和300mTorr之间的压力在-40℃和-10℃之间的温度下应用真空;f.以0.3℃/min至3℃/min增加温度至35℃和50℃之间;和g.等温保持至少10小时或直到残余的水分不超过2%。在另一个实施方案中,水性溶液的冻干包括以下步骤:a.将水性溶液以0.5℃/min至1℃/min的速率冷却至-40℃或更低;b.等温保持至少120min;c.以0.5℃/min至3℃/min的速率升温至-20℃和-15℃之间;d.等温保持至少180min;e.使用50mTorr和200mTorr之间的压力在-30℃和-10℃之间的温度下应用真空;f.以0.5℃/min至1℃/min的速率增加温度至35℃和50℃之间;和g.等温保持至少10小时。在一个实施方案中,冷却步骤a)通过以至少0.3℃/min,例如0.4、0.5、0.6、0.7、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2.0、2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.9或3.0℃/min冷却水性溶液至-40℃的温度进行。在另一实施方案中,冷却步骤a)通过以至少0.3℃/min,例如0.4、0.5、0.6、0.7、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2.0、2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.9或3.0℃/min冷却水性溶液至-50℃的温度进行。在另一实施方案中冷却步骤a)通过以至少0.3℃/min,例如0.4、0.5、0.6、0.7、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2.0、2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.9或3.0℃/min冷却水性溶液至-60℃的温度进行。在另一实施方案中升温步骤c)通过以至少0.3℃/min,例如0.4、0.5、0.6、0.7、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2.0、2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.9、3.0、3.5、3.8、3.9、4.0、4.5、5.0或6.0℃/min将物质升温至-15℃的温度进行。在另一实施方案中升温步骤c)通过以至少0.3℃/min,例如0.4、0.5、0.6、0.7、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2.0、2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.9、3.0、3.5、3.8、3.9、4.0、4.5、5.0或6.0℃/min将物质升温至-20℃的温度进行。在另一实施方案中步骤f)的温度增加以至少0.3℃/min、例如0.4、0.5、0.6、0.7、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2.0、2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.9或3.0℃/min进行至35℃的温度。在另一实施方案中步骤f)的温度增加以至少0.3℃/min、例如0.4、0.5、0.6、0.7、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2.0、2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.9或3.0℃/min进行至40℃的温度。在另一实施方案中步骤f)的温度增加以至少0.3℃/min、例如0.4、0.5、0.6、0.7、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2.0、2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.9或3.0℃/min进行至50℃的温度。用途在另一方面,本发明提供本文任何方面或实施方案中定义的ADC制剂,用于治疗癌症。示例性的癌症包括但不限于,由中枢神经系统的肿瘤引起的那些癌症、头颈癌、肺癌例如NSCLC、乳腺癌、尤其是三阴性乳腺癌、食管癌、胃癌、肝和胆癌、胰腺癌、结肠直肠癌、膀胱癌、肾癌、前列腺癌、子宫内膜癌、卵巢癌、恶性黑素瘤、肉瘤、未知原发来源的肿瘤、骨髓癌、急性成淋巴细胞性白血病、慢性成淋巴细胞性白血病和非霍奇金淋巴瘤、皮肤癌、神经胶质瘤、脑癌、子宫癌、急性髓细胞样白血病和直肠癌。在一个实施方案中,ADC的抗体部分是抗-TF抗体并且将本发明的制剂给予患有胰腺癌、结肠直肠癌、乳腺癌、膀胱癌、前列腺癌或卵巢癌的受试者。在给予受试者之前,将本发明的包含治疗有效量的ADC的冻干制剂在药学上可接受的稀释剂中溶解,即重构。示例性的非限制性稀释剂包括无菌药用级水(注射用水,WFI)或盐水、抑菌注射用水(BWFI)、pH缓冲溶液(例如磷酸盐-缓冲盐水)、林格溶液和葡萄糖溶液。例如,本发明的冻干制剂可在水、pH缓冲溶液(例如磷酸盐-缓冲盐水)、林格溶液和葡萄糖溶液中重构。例如,本发明的冻干制剂可在无菌注射用水(WFI)中重构至约5至约30mg/mLADC、例如约7至约20mg/mLADC、例如约8至15mg/mLADC、例如约9至约11mg/mLADC、例如约10mg/mLADC的浓度。浓缩物可任选地进一步稀释,用于例如注入pH缓冲溶液(例如磷酸盐-缓冲盐水、林格溶液和/或葡萄糖溶液)至约0.05mg/mL至30mg/mLADC、例如0.12mg/mL至2.40mg/mLADC的浓度。通常,本发明的重构制剂适合于胃肠外给予。本文使用的词语"胃肠外给予"和"经胃肠外给予"意指并非肠内和局部给予的给予方式,通常通过注射或输注,并且包括表皮、静脉内、肌内、动脉内、鞘内、囊内、眶内、心内、皮内、腹膜内、腱内、经气管、皮下、表皮下、关节内、被膜下、蛛网膜下、脊柱内、颅内、胸内、硬膜外和胸骨内注射和输注。在一个实施方案中,药物组合物通过静脉内或皮下注射或输注给予,例如通过在无菌水或盐水中重构冻干制剂,和保持在IV袋或注射器中,然后给予受试者。本发明还提供包含本发明的抗-TFADC的冻干制剂的药盒,通常在密封容器中,例如小瓶、安瓿或小药囊(sachette),指示活性剂的数量。在药物通过注射给予时,可提供例如无菌注射用水或盐水的安瓿,任选地作为药盒的一部分,使得各成分可在给予之前混合。这样的药盒可进一步包括(如果需要的话)各种常规药盒组分中的一种或多种,例如含有一种或多种药学上可接受的载体的容器、另外的容器等等,如本领域技术人员显而易见的。作为插页或作为标签指示待给予的组分的数量、给药指南和/或用于混合各组分的指南的印刷说明书,也可包括在药盒中。本发明的实施方案:1.抗-组织因子(TF)抗体-药物缀合物(ADC)的冻干制剂,所述冻干制剂可通过冻干包含约5g/L至约30g/L抗-TFADC和药学上可接受的赋形剂的水性制剂获得或通过冻干包含约5g/L至约30g/L抗-TFADC和药学上可接受的赋形剂的水性制剂获得,所述赋形剂包含:a.具有约5至约7的pH的约20至约50mM组氨酸或柠檬酸盐缓冲液;b.约10至约250mM蔗糖或海藻糖;和c.约50mM至约300mM甘露醇或甘氨酸。2.抗-TFADC的冻干制剂,所述冻干制剂可通过冻干水性制剂获得或通过冻干水性制剂获得,所述水性制剂包含a.抗-TFADC,其包含选自MMAE和MMAF的治疗部分和包含选自以下的VH和VL区的抗-TF抗体:包含SEQIDNO:5的氨基酸序列的VH区和包含SEQIDNO:45的氨基酸序列的VL区;包含SEQIDNO:33的氨基酸序列的VH区和包含SEQIDNO:73的氨基酸序列的VL区;包含SEQIDNO:37的氨基酸序列的VH区和包含SEQIDNO:77的氨基酸序列的VL区;或包含SEQIDNO:1的氨基酸序列的VH区和包含SEQIDNO:41的氨基酸序列的VL区;和b.药学上可接受的赋形剂,包含:限制在冻干步骤期间的pH变化的缓冲液;至少一种非还原糖,其与固体状态的抗-TFADC形成非晶相;和至少一种填充剂,其中所述冻干制剂基本上不含任何表面活性剂。3.实施方案2的冻干制剂,其中所述水性制剂包含选自组氨酸、柠檬酸盐、磷酸盐、碳酸、琥珀酸盐、乙醇酸盐和其任何的组合的缓冲液。4.实施方案1或3中任一个的冻干制剂,其中缓冲液是组氨酸。5.前述实施方案中任一个的冻干制剂,其中水性制剂包含约7至约20g/L抗-TFADC,例如约8至约15g/L抗-TFADC。6.前述实施方案中任一个的冻干制剂,其中水性制剂包含约9至约11g/L抗-TFADC。7.前述实施方案中任一个的冻干制剂,其中水性制剂的pH在约5.5至6.5的范围内,例如约6。8.前述实施方案中任一个的冻干制剂,其中所述水性制剂包含约25至约40mM组氨酸,例如约28至约34mM组氨酸。9.前述实施方案中任一个的冻干制剂,其中所述水性制剂包含约29至约31mM组氨酸。10.前述实施方案中任一个的冻干制剂,其中所述水性制剂包含约50至约225mM蔗糖或海藻糖,例如84至约165mM蔗糖或海藻糖。11.前述实施方案中任一个的冻干制剂,其中所述水性制剂包含约84至约146mM蔗糖,例如约84至约92mM蔗糖。12.前述实施方案中任一个的冻干制剂,其中所述水性制剂包含约100至约274mM甘露醇或甘氨酸。13.前述实施方案中任一个的冻干制剂,其中所述水性制剂包含约158至约172mM甘露醇。14.前述实施方案中任一个的冻干制剂,其中所述水性制剂包含约9至约11g/L抗-TFADC,例如约10mg/mL抗-TFADC,和约30mM组氨酸,约88mM蔗糖和约165mM甘露醇。15.冻干制剂,其包含a.抗-TF抗体药物缀合物,b.缓冲剂,选自组氨酸、柠檬酸盐、琥珀酸盐、乙醇酸盐、碳酸和磷酸盐;c.非还原糖,选自蔗糖、海藻糖和其组合;和d.填充剂,选自甘露醇和甘氨酸。16.前述实施方案中任一个的冻干制剂,其基本上不含任何聚山梨醇酯。17.前述实施方案中任一个的冻干制剂,其基本上不含任何表面活性剂。18.前述实施方案中任一个的冻干制剂,其不含任何表面活性剂。19.冻干制剂,其基本上由以下组成:a.抗-TF抗体药物缀合物,b.缓冲剂,选自组氨酸和柠檬酸盐;c.非还原糖,选自蔗糖、海藻糖和其组合;和d.填充剂,选自甘露醇和甘氨酸。20.前述实施方案中任一个的冻干制剂,其包含甘露醇和蔗糖,其中甘露醇与蔗糖的重量比为至少约1。21.前述实施方案中任一个的冻干制剂,其包含甘露醇和蔗糖,其中甘露醇与蔗糖的重量比在约1和约30之间,例如在1和约10之间,例如在约1和约2之间。22.前述实施方案中任一个的冻干制剂,包含甘露醇和蔗糖,其中甘露醇与蔗糖的重量比为约1。23.前述实施方案中任一个的冻干制剂,包含甘露醇,其中甘露醇与抗-TFADC的重量比为至少约3。24.前述实施方案中任一个的冻干制剂,包含甘露醇和蔗糖,其中甘露醇与抗-TFADC的重量比为约3和甘露醇与蔗糖的重量比为约1。25.前述实施方案中任一个的冻干制剂,其可通过冻干水性制剂获得或通过冻干水性制剂获得,所述水性制剂包含少于约250mM蔗糖和少于约300mM甘露醇。26.前述实施方案中任一个的冻干制剂,其可通过冻干水性制剂获得或通过冻干水性制剂获得,所述水性制剂包含少于约160mM蔗糖和少于约274mM甘露醇。27.前述实施方案中任一个的冻干制剂,其可通过冻干水性制剂获得或通过冻干水性制剂获得,所述水性制剂包含约7至约20g/L抗-TFADC和a.约28至34mM组氨酸;b.约84至约146mM蔗糖;c.约158至约274甘露醇;或d.(a)至(c)的任何两个或全部的组合。28.前述实施方案中任一个的冻干制剂,其可通过冻干水性制剂获得或通过冻干水性制剂获得,所述水性制剂包含约9至约11g/L抗-TFADC,例如约10mg/mL抗-TFADC,和a.约30mM组氨酸;b.约88mM蔗糖;c.约165mM甘露醇;或d.(a)至(c)的任何两个或全部的组合。29.前述实施方案中任一个的冻干制剂,其中每抗体分子的药物部分的平均绝对数是1、2、3、4、5、6、7或8,例如3、4或5。30.前述实施方案中任一个的冻干制剂,其中抗体与疏水性药物缀合。31.前述实施方案中任一个的冻干制剂,其中药物是奥里斯他汀或其功能性肽类似物或衍生物。32.前述实施方案中任一个的冻干制剂,其中药物与抗-TF抗体经过连接至抗-TF抗体的巯基残基的接头连接,所述巯基残基通过部分还原或还原抗-TF抗体获得。33.前述实施方案中任一个的冻干制剂,其中药物是MMAE或MMAF。34.前述实施方案中任一个的冻干制剂,其中药物与抗-TF抗体经过接头-药物连接,所述接头-药物选自:vcMMAE、vcMMAF或mcMMAF。35.前述实施方案中任一个的冻干制剂,其中抗体是全长抗体。36.前述实施方案中任一个的冻干制剂,其中抗体是人IgG1κ抗体。37.前述实施方案中任一个的冻干制剂,其中抗体与一种或多种参比抗体竞争结合人TF,所述参比抗体包含选自以下的VH和VL区:a.包含SEQIDNO:5的氨基酸序列的VH区和包含SEQIDNO:45的氨基酸序列的VL区;b.包含SEQIDNO:33的氨基酸序列的VH区和包含SEQIDNO:73的氨基酸序列的VL区;c.包含SEQIDNO:37的氨基酸序列的VH区和包含SEQIDNO:77的氨基酸序列的VL区;或d.包含SEQIDNO:1的氨基酸序列的VH区和包含SEQIDNO:41的氨基酸序列的VL区。38.前述实施方案中任一个的冻干制剂,其中抗-TF抗体包含a.可变重链(VH)区,包含具有SEQIDNO:6所示的氨基酸序列的CDR1区、具有SEQIDNO:7所示的氨基酸序列的CDR2区和具有SEQIDNO:8所示的氨基酸序列的CDR3区,和可变轻链(VL)区,包含具有SEQIDNO:46所示的氨基酸序列的CDR1区、具有SEQIDNO:47所示的氨基酸序列的CDR2区和具有SEQIDNO:48所示的氨基酸序列的CDR3区;b.VH区,包含具有SEQIDNO:34所示的氨基酸序列的CDR1区、具有SEQIDNO:35所示的氨基酸序列的CDR2区和具有SEQIDNO:36所示的氨基酸序列的CDR3区,和VL区,包含具有SEQIDNO:74所示的氨基酸序列的CDR1区、具有SEQIDNO:75所示的氨基酸序列的CDR2区和具有SEQIDNO:76所示的氨基酸序列的CDR3区,或者c.VH区,包含具有SEQIDNO:38所示的氨基酸序列的CDR1区、具有SEQIDNO:39所示的氨基酸序列的CDR2区和具有SEQIDNO:40所示的氨基酸序列的CDR3区,和VL区,包含具有SEQIDNO:78所示的氨基酸序列的CDR1区、具有SEQIDNO:79所示的氨基酸序列的CDR2区和具有SEQIDNO:80所示的氨基酸序列的CDR3区,或者d.VH区,包含具有SEQIDNO:2所示的氨基酸序列的CDR1区、具有SEQIDNO:3所示的氨基酸序列的CDR2区和具有SEQIDNO:4所示的氨基酸序列的CDR3区,和VL区,包含具有SEQIDNO:42所示的氨基酸序列的CDR1区、具有SEQIDNO:43所示的氨基酸序列的CDR2区和具有SEQIDNO:44所示的氨基酸序列的CDR3区,或者e.任何所述抗体的变体,其中所述变体优选地在所述序列中具有至多1、2或3个氨基酸修饰,更优选地氨基酸取代,例如保守氨基酸取代。39.前述实施方案中任一个的冻干制剂,其中抗体包含a.包含SEQIDNO:5的氨基酸序列的VH区和包含SEQIDNO:45的氨基酸序列的VL区;b.包含SEQIDNO:33的氨基酸序列的VH区和包含SEQIDNO:73的氨基酸序列的VL区;c.包含SEQIDNO:37的氨基酸序列的VH区和包含SEQIDNO:77的氨基酸序列的VL区;或d.包含SEQIDNO:1的氨基酸序列的VH区和包含SEQIDNO:41的氨基酸序列的VL区。40.前述实施方案中任一个的冻干制剂,其中抗体是全长的完全人单克隆IgG1抗体,任选地IgG1κ。41.适合于制备抗-TFADC的冻干制剂的水性溶液,包含a.约7至约20g/L抗-TFADC,任选地其中抗体部分包含实施方案37至39中任一个的VH和VL序列,b.约28至34mM组氨酸;c.约84至约146mM蔗糖;d.约158至约274甘露醇;或e.(b)至(d)中任何两个或全部的组合。42.药学上可接受的液体制剂,其通过在无菌水性稀释剂中重构实施方案1-40中任一个的冻干制剂获得。43.实施方案42的液体制剂,其包含约5g/L至约30g/L抗-TFADC、具有约5至约7的pH的约20至约50mM组氨酸、约10至约250mM蔗糖或海藻糖、和约50mM至约300mM甘露醇或甘氨酸。44.实施方案43的液体制剂,其包含约9至约11mg/mL抗-TFADC、约28至约34mM组氨酸、约84至约92mM蔗糖和约158至约274mM甘露醇。45.制备抗-TFADC的可注射溶液的方法,包括在无菌水性稀释剂中重构实施方案1-40中任一个的冻干制剂的步骤。46.抗-TFADC的冻干制剂,所述冻干制剂通过冻干水性制剂制备,所述水性制剂包含约9g/L至约11g/L抗-TFADC和药学上可接受的赋形剂,所述赋形剂包含:具有约5至约7的pH的约30mM组氨酸缓冲液;约88mM蔗糖;和约165mM甘露醇;其中抗体包含包含SEQIDNO:5的氨基酸序列的VH区和包含SEQIDNO:45的氨基酸序列的VL区;包含SEQIDNO:33的氨基酸序列的VH区和包含SEQIDNO:73的氨基酸序列的VL区;包含SEQIDNO:37的氨基酸序列的VH区和包含SEQIDNO:77的氨基酸序列的VL区;或包含SEQIDNO:1的氨基酸序列的VH区和包含SEQIDNO:41的氨基酸序列的VL区,和其中接头-药物是vcMMAE、vcMMAF或mcMMAF。实施例1本实施例显示某些缓冲液和pH值对抗-TFHuMab098(参见WO2011157741A2,本文称为HuMax-TF)的热稳定性的作用。选择的赋形剂、缓冲液和pH条件的加速稳定性研究使用pH4.5-6.5的组氨酸和乙酸盐缓冲液系统进行设计。使用的DOE(实验设计)为反应表面、线性设计,具有一个数字(pH)和两个类别因素(缓冲液和赋形剂类型)。对于各制剂组合物,DOE包括重复的样品以评价与各种生物物理和分析方法有关的实验变异性。另外的在中心点制剂中50mg/mL和具有组合的山梨醇和氯化钠赋形剂的20mg/mL的偏离-DOE(off-DOE)的样品包括在研究中。表2显示HuMax-TF的预制剂实验设计(DOE)样品。表2加速稳定性样品通过各种分析方法测试。选择的结果在实施例2、3和4中讨论。实施例2在5和45℃(分别为无应激和应激)孵育4周的HuMax-TF样品使用SEC(大小排阻色谱)进行分析以确定各种制剂对聚集和降解的程度的影响。无应激HuMax-TF样品的SEC色谱显示一个主峰,其对应于单体抗体,占总峰面积的超过98%。该一个主要的SEC峰反映了各制剂中抗体的大小均匀性。为比较各种制剂之间的SEC数据,将在主峰之前洗脱的各物质的峰面积值组合和报告为百分比HMW。类似地,将在主峰之后洗脱的各物质的峰面积值组合和报告为百分比降解。对于在未应激条件下任何制剂的任何物质,图1提供的条柱图未显示显著趋势。然而,对于应激条件,由于聚集和降解导致,观察到%HMW和%LMW增加,和%主峰值降低。相对于组氨酸制剂,对于乙酸盐制剂观察到较高的%HMW和%LMW值。对于两种缓冲液类型,和在乙酸盐制剂中更加显著,%HMW和%LMW物质随着pH增加而降低,并且相对于氯化钠,用山梨醇而降低。实施例3对选择的4周DOE样品,在还原和非还原条件下进行SDS-PAGE分析。在还原条件下,重链条带出现在大约54kDa,而轻链条带出现在大约26kDa。在非还原条件下,主要的HuMax-TF条带迁移至大约145kDa。样品使用还原和非还原SDS-PAGE分析。与未应激样品相比,热应激的制剂显示在非还原凝胶中在完整IgG下面的LMW条带的强度增加。类似地,相对于未应激样品,对于应激样品,在重链和轻链条带之间观察到新条带的强度增加。对于还原和非还原样品两者,相对于组氨酸制剂,乙酸盐制剂显示这些降解物质的强度的更大增加。实施例4使用cIEF(成像毛细管等电聚焦)分析1周应激和未应激HuMax-TFDOE样品,以评价各种制剂组分对观察到的峰面积的影响。结果显示在图2中。将对于具有低于主峰pI值的pI值的各物质的百分比峰面积值合并和报告为百分比酸性,和将对于具有高于主峰pI值的pI值的各物质的百分比峰面积值合并和报告为百分比碱性。图2中提供的未应激HuMax-TFDOE样品的cIEF分析未显示,对于未应激样品,cIEF结果对pH、缓冲液类型或赋形剂类型的依赖性的任何明显的趋势。应激DOE样品的cIEF分析显示主峰百分比面积的5-13%降低。主峰面积的降低伴随有酸性物质的峰面积增加,并且对于大多数制剂,百分比碱性峰面积也增加。在条柱图中描述的百分比酸性物质的变化未显示明显的pH依赖性,但乙酸盐制剂普遍显示比组氨酸制剂略微更低的百分比酸性峰面积。然而,百分比碱性物质显示强的pH依赖性,因为百分比碱性峰面积随pH增加而降低。另外,组氨酸制剂显示比乙酸盐制剂更低的百分比碱性物质。实施例5筛选研究经设计以测试pH、聚山梨醇酯的存在和山梨醇的存在对溶液中的HuMax-TF-ADC的稳定性的影响。HuMax-TF-ADC是由人单克隆IgG1抗体HuMax-TF011通过蛋白酶可裂解缬氨酸瓜氨酸(vc)接头经化学缀合至微管破坏剂单甲基奥里斯他汀E(MMAE)构成的抗体-药物缀合物。12种不同的制剂使用3个不同的pH值制备。溶液制剂研究设计显示在表3中。所有制剂包含10mg/mL的HuMax-TF-ADC与30mM组氨酸。Tween=聚山梨醇酯80(PS80)。表3制剂pHTween浓度(%)山梨醇(mM)15.50025.5022535.50.02045.50.0222556.00066.0022576.00.02086.00.0222596.500106.50225116.50.020126.50.02225制备用于筛选研究的12种制剂使用UV/Vis(紫外-可见光谱法)、icIEF、SEC和DAR-HIC(药物与抗体摩尔比,使用疏水性相互作用色谱)进行分析。制剂筛选研究已经显示,HuMax-TF-ADC的主峰稳定性指示参数是通过SEC测量的聚集体和通过icIEF测量的酸性同种型(推测归因于脱酰胺)。pH对在40℃贮存2周的溶液样品中HMW形成的百分数的影响通过SEC测量(图3显示表3的制剂1、5和9的SEC%HMW)。6.0的pH显示对限制组氨酸缓冲液中HuMax-TF-ADC聚集是最有效的。pH对在40℃贮存2周的溶液样品中HuMax-TF-ADC的脱酰胺的影响通过icIEF中的酸性物质增加来测量(图4显示表3的制剂1、5和9的结果)。发现pH5.5对限制脱酰胺是最有效的,接着是pH6.0和pH6.5。在40℃贮存2周的HuMax-TF-ADC制剂中,使用山梨醇或PS80对通过icIEF的百分比主料峰以及通过SEC的主峰不具有任何显著影响(图5A和5B)。这表明制剂可不含表面活性剂。实施例6制剂筛选已表明,主峰稳定性指示参数是通过SEC测量的聚集体和通过icIEF测量的酸性同种型(推测为脱酰胺)。在所有测试的液体制剂中在加速条件下测量的脱酰胺速率是快速的,因此HuMax-TF-ADC的测试的液体制剂明显是不稳定的。在一些液体制剂和初步的冻干制剂之间的比较示于图6。实施例7试验性的冻干研究用包含不同浓度的蔗糖(150mM和250mM)、10mg/mLHuMax-TF-ADC/30mM组氨酸缓冲液pH6.0的两种制剂进行,以研究填料体积和小瓶体积/形状对冻干结块的可能影响。对于HuMax-TF-ADC的组氨酸、蔗糖制剂,冻干结块外观由于不同的填料体积和小瓶大小而不同:A:将第一组制剂样品装入0.25mL/2mL玻璃小瓶和冻干。样品显示药学上精致性,没有明显的塌陷迹象。B:将相同制剂的第二组样品使用4mL填料体积在10mL小瓶中同时冻干,使用相同的冻干循环。样品显示严重皱缩。因此,对于低填料体积0.25ml/2mL玻璃小瓶,制剂可能支持药学精致的结块,但较不适合于更高填料体积4mL/10mL小瓶。实施例8本实施例的目的是测试蔗糖和甘露醇浓度对HuMax-TF-ADC的冻干制剂的稳定性的影响。最初的测试显示,蔗糖可容易地用海藻糖置换,保持制剂的主要性质。测试仅使用蔗糖继续。包含10mg/mLHuMax-TF-ADC、30mM组氨酸pH6.0的三种不同的冻干制剂用不同浓度的蔗糖和甘露醇制备。细节显示在表4中。表4制剂HuMax-TF-ADC组氨酸蔗糖甘露醇A10mg/mL30mM225mM(7.7%w/v)274mM(5%w/v)B10mg/mL30mM88mM(3%w/v)165mM(3%w/v)C10mg/mL30mM160mM(5.5%w/v)274mM(5%w/v)在25℃、40℃和50℃,将具有不同浓度的蔗糖和甘露醇的冻干制剂置于加速稳定性测试中。应激样品通过方法例如SEC(大小排阻色谱)、icIEF(成像毛细管等电聚焦)、FTIR(傅里叶变换红外光谱)、DLS(动态光散射)进行分析。3种制剂的加速稳定性数据表明,关于聚集体和主料同种型,制剂B相对于其它制剂具有优势,尤其是当比较在50℃和40℃的稳定性数据时。示例性的结果显示在图7中。此外,当与制剂A和C相比时,在40℃贮存2个月后制剂B在更多微粒中显示最小增长(图8)。在50℃贮存2周后,在制剂A、B和C的第二衍生物FTIR光谱的酰胺I区中未观察到显著变化(图9)。数据表明当在应激温度条件下贮存时,分子的第二结构在各制剂中保持相同。实施例11该实施例验证甘露醇作为HuMax-TF-ADC制剂中的结晶填充剂的合适性,尤其是在HuMax-TF-ADC的冻干制剂中甘露醇结晶的退火温度和时间。差示扫描量热法(DSC)是直接测量能量和允许精确度量样品的热容量的热分析技术。当样品经历物理转化,例如相转变时,样品和参比之间的热流差异可通过DSC检测。在本实施例中,DSC用于测定制剂中甘露醇开始结晶所需要的时间。测试了两个不同的退火温度(-15℃和-20℃)。在溶液以1℃/min冷却至-40℃时,温度以1℃/min匀变至-15℃或-20℃,和等温保持120分钟。当在-15℃或-20℃退火时,制剂B的甘露醇结晶的开始时间为大约10分钟,如在图10中所示。数据证实,在退火期间甘露醇容易结晶,因此充分起到制剂的结晶填充剂的作用。实施例10以长期和加速稳定性程序评价冻干HuMaxTF-ADC制剂样品。在重构后HuMax-TF-ADC制剂的组成是10mg/mL,其在30mM组氨酸(对应于4.65mg/mL)、88mM蔗糖(对应于大约30mg/mL)、165mM甘露醇(对应于大约30mg/mL)pH6.0中配制。长期稳定性样品在以下时间进行分析:零、1个月、2个月、3个月、6个月、9个月、12个月和18个月等等。在不同的时间点,将冻干制剂用注射用水(WFI)重构和通过分析方法包括SEC、HIC、CE-SDS和icIEF进行测试。在5±3℃和25±2℃至少6个月贮存后,所有样品通过所有测试方法保持稳定。与研究开始相比,通过任何测试方法,在5±3℃和25±2℃贮存的样品在至少6个月时间点未显示显著变化。在25℃加速稳定性测试期间通过icIEF、还原CE-SDS和SEC测试,观察到纯度特征的预期较少变化。具体而言,关于聚集,SEC分析显示对于在5±3℃和25±2℃两者经过至少6个月的样品,聚集体的百分数保持在大约2.2%。IcIEF用于测定HuMax-TF-ADC的载料特征的变化。与时间零相比,经过至少6个月,对于在5±3℃贮存的样品,主料同种型的变化在0.4%内,和对于在25℃贮存的样品,变化仅为1.7%。此外,在5℃和25℃两者经过至少6个月,平均DAR(摩尔奥里斯他汀/摩尔mAb)、药物载量和游离药物几乎保持恒定。在5℃和25℃两者经过至少6个月没有降解迹象,如通过CE-SDS(非还原)和CE-SDS(还原)所示,以及通过SEC的LMW。最后但非最不重要,通过细胞毒性的生物测定证明,HuMaxTF-ADC的生物功能可在5℃和25℃两者下保持至少6个月。因此,在这样的制剂中HuMax-TF-ADC对于药物应用具有可接受的稳定性。表5A:在5±3℃来自HuMax-TF-ADC药物产物的稳定性程序的实施例数据CR=与参比标准相当;HC=重链;LC=轻链;TQI=总可计量杂质;NT=未测试;NA=未分析表5B:在25±3℃来自HuMax-TF-ADC药物产物的稳定性程序的实施例数据CR=与参比标准相当;HC=重链;LC=轻链;TQI=总可计量杂质;NT=未测试;NA=未分析实施例11本研究检查较高或较低赋形剂浓度对HuMax-TF-ADC的稳定性的影响,以验证赋形剂浓度的可接受范围。三种HuMax-TF-ADC制剂根据表6制备。将溶液以4mL/瓶装入10mL玻璃小瓶和冻干。将各制剂的样品置于50℃加速稳定性4周,和在2、3和4周后取样。测试样品的浓度、pH、重构时间、外观、iCE、CE-SDS、SEC、DAR-HIC、游离药物DLS和MFI,以获得数据来评价较高或较低浓度的赋形剂对HuMax-TF-ADC的稳定性的影响。数据表明,制剂在应激稳定性测试中行为类似。表6实施例12检查了HuMax-TF-ADC浓度对冻干HuMax-TF-ADC制剂的稳定性的影响,以验证制剂中HuMax-TF-ADC浓度的可接受范围。将通过30mM组氨酸、88mM蔗糖、165mM甘露醇和5mg/mL或30mg/mLHuMax-TF-ADC制备的制剂冻干和在40℃贮存2个月或在50℃贮存2周。在40℃2个月贮存后通过SEC的主峰停留在97%以上(图12),对于两个浓度在40℃贮存后仅观察到平均百分比高分子量物质的略微增加(图13)。在应激条件下载料特征变化的趋势,如通过ICE数据所示,对于两个浓度是类似的(图14、15和16)。这表明,HuMax-TF-ADC可至少在5mg/mL和30mg/mL的范围中的浓度下配制。实施例13HuMax-TF-ADC的使用中稳定性以不同浓度(至多48mg/ml)和不同稀释剂即注射用水(WFI)、0.9%NaCl(盐水)和葡萄糖5%(D5W)溶液在室温下进行研究至少48小时。SEC数据表明,对于在25℃贮存48小时的样品,平均百分比主峰保持大于97%(图17)。48mg/mL样品包含比其它样品多大约0.5%的高分子量物质(图18)。不同的稀释剂未影响聚集倾向。还使用iCE检查了在25℃贮存的使用中溶液样品。当与其它样品相比,用5%葡萄糖重构的样品具有在48小时后最低的百分比主峰(图19)和最高的百分比酸性物质(图20)。这表明对于保持HuMax-TF-ADC的载料特征,WFI和盐水可能是更好的稀释剂。实施例14本实施例证实了缓冲液类型和结晶赋形剂类型对HuMax-TF-ADC制剂的稳定性的影响。当用柠檬酸盐替代组氨酸作为冻干制剂中的缓冲液时,或当用甘氨酸替代甘露醇作为结晶赋形剂时,在40℃下贮存2个月后它们的稳定性通过SEC和iCE进行分析。在图22至图26中,图标“甘氨酸”是指含10mg/mLHuMax-TF-ADC、30mM组氨酸、88mM蔗糖、165mM甘氨酸pH6.0的制剂;而图标“柠檬酸盐”是指含10mg/mLHuMax-TF-ADC、30mM柠檬酸盐、88mM蔗糖、165mM甘露醇pH6.0的制剂。在40℃下贮存2个月后在柠檬酸盐或甘氨酸制剂中未观察到SEC平均百分比主峰(图22)或高分子峰(图23)的显著变化。在任何样品中未观察到低分子峰。在含甘氨酸和柠檬酸盐的制剂中在40℃2个月时iCE百分比主峰的降低率(图24)与含30mM组氨酸、88mM(3%)蔗糖和165mM(3%)甘露醇的制剂(图7)相当。观察到,在热应激条件下,与具有组氨酸的制剂相比,柠檬酸盐制剂与更多酸性物质有关(图25),而与具有甘露醇的制剂相比,甘氨酸制剂与更多碱性物质有关(图26)。实施例15本实施例证实了pH和缓冲液浓度对HuMax-TF-ADC的稳定性的影响。HuMax-TF-ADC以10mg/mL浓度用3%甘露醇和3%蔗糖,但用20mM或50mM组氨酸制备,将样品的pH调整至5、6或7。将冻干样品在40℃下贮存至多2个月。不管缓冲液浓度和pH如何,用20mM组氨酸或50mM组氨酸在pH5、6或7下制备的制剂在40℃显示类似的SEC结果,除了具有50mM组氨酸在pH5的制剂显示聚集的略微增加之外(图30和31)。如图27-29中通过iCE所示的,观察到关于在应激测试下载料特征变化,pH比缓冲液浓度起更重要的作用。在pH6制备的制剂显示在40℃贮存2个月期间主峰的最少降低。观察到主峰明显降低,其可能大于分析方法变化。在pH5制备的制剂显示百分比主峰的最大降低(图27)。在pH7制备的制剂具有最高的酸性物质百分数(图28)和在pH5制备的制剂具有最高的碱性物质百分数(图29)。实施例16本发明的HuMax-TF-ADC的冻干制剂的冻干循环可如下所述进行。首先,可将小瓶以0.5℃/min至1℃/min冷却至-40℃或更低,和等温保持至少120min(冷却步骤)。随后将小瓶以0.5℃/min至3℃/min的速率升温至-20℃和-15℃之间和等温保持至少180min(退火步骤)。之后,使用50mTorr和200mTorr之间的压力用-30℃和-10℃之间的温度开始真空(初级干燥)。最后,温度以0.5℃/min至1℃/min增加至35℃和50℃之间和等温保持至少10小时(次级干燥)。残留的水分不应超过2%重量。参考文献列表WO2011157741、WO9704801、WO9856418、WO02011753、WO02096457、WO03009817、WO03039485、US8372396、WO2004004639、WO2004016286、WO2004055164、WO2004071439、WO2006014965、WO2006044908和WO2007019232。PhysicochemicalStabilityoftheAntibody−DrugConjugateTrastuzumab-DM1:ChangesduetoModificationandConjugationProcesses.AdityaA.Wakankar等,BioconjugateChemistry2010:21(9),1588-1595Challengesindevelopingbioanalyticalassaysforcharacterizationofantibody-drugconjugates.StephanJP等,Bioanalysis.2011Mar;3(6):677-700Analyticalmethodsforphysicochemicalcharacterizationofantibodydrugconjugates.WakankarA等,MAbs.2011Mar-Apr;3(2):161-72.Analyticalandbioanalyticaltechnologiesforcharacterizingantibody–drugconjugates,StephenCAlley,KevinEAnderson,CurrentOpinioninChemicalBiology,June2013,17(3),406-411EffectofPolysorbate80QualityonPhotostabilityofaMonoclonalAntibody,Singh等,AAPSPharmSciTech,Vol.13,No.2,2012.Polysorbates20and80UsedintheFormulationofProteinBiotherapeutics:StructureandDegradationPathways.BRUCEA.KERWIN,JOURNALOFPHARMACEUTICALSCIENCES,VOL.97,NO.8,AUGUST2008,2924-2935.Aggregatesinmonoclonalantibodymanufacturingprocesses,Vásquez-Rey和Lang,2011,Biotech,Bioeng.108(7)p.1494-1508.当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1