抗真菌干燥粉末的制作方法
本申请要求2016年10月14日提交的美国专利申请号62/408,376的权益,该专利申请全部内容以引用的方式并入本文。
背景技术:
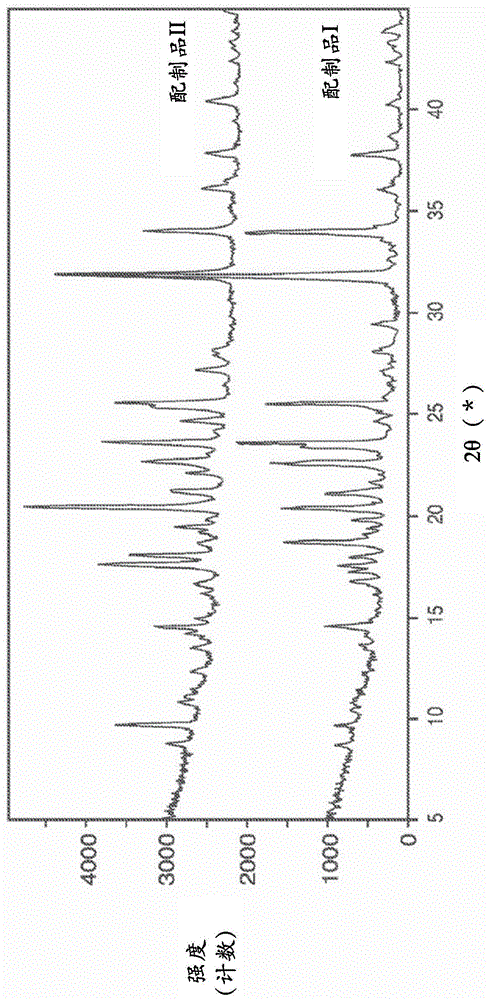
:由曲霉属(aspergillusspp.)和其他真菌导致的肺部真菌感染在呼吸功能下降的患者(如囊性纤维化(cf)患者)中越来越受到关注。例如,患者可能患有慢性肺部真菌感染或变应性支气管肺曲霉菌病(abpa),这是一种严重的炎症病状,典型地用长期口服类固醇治疗。许多抗真菌剂是已知的,包括三唑(例如伊曲康唑)、多烯(例如两性霉素b)、和棘白菌素。抗真菌剂典型地具有低水溶性和差的口服生物利用度,并且获得可以给予以提供安全和治疗水平的抗真菌剂的药物配制品一直是具有挑战性的。抗真菌剂典型地作为口服或静脉内(iv)配制品给予,作为真菌感染(包括肺部感染和abpa)的治疗。然而,此类配制品受到差的口服生物利用度、不利的副作用和毒性、以及广泛的药物-药物相互作用的限制。替代方法(如通过吸入递送到气道)理论上可以减少全身副作用也存在挑战。值得注意地,熟知的是水溶性差的药剂在吸入时产生局部肺毒性(例如局部炎症、肉芽肿)。解决难溶性药剂局部毒性的常规方法是例如使用无定形配制品配制该药剂以提高其溶解速率。伊曲康唑的化学结构描述于美国专利号4,916,134中。伊曲康唑是一种三唑抗真菌剂,其提供治疗益处(例如,用于治疗真菌感染),并且是可以口服或静脉内递送的(伊曲康唑;詹森制药公司(janssenpharmaceuticals))中的活性成分。伊曲康唑可以使用本领域熟知的各种方法合成。需要可以安全地给予以治疗真菌感染的抗真菌剂的新配制品。技术实现要素:本发明涉及包含均匀的可吸入干燥颗粒的干燥粉末配制品,这些均匀的可吸入干燥颗粒含有1)呈结晶微粒形式的抗真菌剂,2)稳定剂,和任选的3)一种或多种赋形剂。在一个具体方面,呈结晶微粒形式的抗真菌剂不是多烯抗真菌剂。在另一个具体方面,本发明涉及1)呈结晶微粒形式的三唑抗真菌剂,2)稳定剂,和任选的3)一种或多种赋形剂。在更具体的方面,三唑抗真菌剂是伊曲康唑。在本发明的所有方面,呈结晶微粒形式的抗真菌剂是约50nm至约5,000nm(dv50)的亚颗粒形式。在一个实施例中,亚颗粒是约50nm至约800nm(dv50)。在一个实施例中,亚颗粒是约50nm至约300nm(dv50)。在一个实施例中,亚颗粒是约50至约200nm(dv50)。在一个实施例中,亚颗粒是约100nm至约300nm(dv50)。抗真菌剂以按重量计约1%至约95%的量存在。抗真菌剂以按重量计约40%至约90%的量存在。抗真菌剂以按重量计约55%至约85%的量存在。抗真菌剂以按重量计约55%至约75%的量存在。抗真菌剂以按重量计约65%至约85%的量存在。抗真菌剂以按重量计约40%至约60%的量存在。优选地,抗真菌剂是至少约50%结晶。抗真菌剂:稳定剂的比率(wt:wt)是从约1:1至50:1。抗真菌剂:稳定剂的比率可以大于或等于10:1,约10:1、或约20:1。抗真菌剂:稳定剂的比率可以是约5:1至约20:1、约7:1至约15:1、或约9:1至约11:1。稳定剂以按重量计约0.05%至约45%的量存在。稳定剂可以以按重量计约4%至约10%的量存在。一种或多种赋形剂以按重量计约10%至约99%的量存在。一种或多种赋形剂可以以按重量计约5%至约50%的量存在。赋形剂可以是钠盐。在一些实施例中,一种或多种赋形剂可以包含一价金属阳离子盐、二价金属阳离子盐、氨基酸、糖醇、或其组合。一种或多种赋形剂可以包含钠盐和氨基酸,如氯化钠或硫酸钠,和亮氨酸。一种或多种赋形剂可以包含镁盐和氨基酸,如乳酸镁和亮氨酸。在一些实施例中,稳定剂可以是聚山梨醇酯80或油酸或其盐。稳定剂可以是聚山梨醇酯80并且以10wt%或更少的量存在。稳定剂可以是聚山梨醇酯80并且以7wt%或更少的量存在。稳定剂可以是聚山梨醇酯80并且以3wt%或更少的量存在。稳定剂可以是油酸并且以10wt%或更少的量存在。稳定剂可以是油酸并且以7wt%或更少的量存在。稳定剂可以是油酸并且以3wt%或更少的量存在。在一些实施例中,可吸入干燥颗粒可以具有约10微米或更小、约5微米或更小的体积中值几何直径(vmgd)。在一些实施例中,可吸入干燥颗粒可以具有0.2g/cc或更大的振实密度。可吸入干燥颗粒可以具有在0.2g/cc与1.0g/cc之间的振实密度。在一些实施例中,干燥粉末具有在约1微米与约5微米之间的mmad。如通过激光衍射测量,干燥颗粒可以具有小于约1.5的1/4巴分散比(1/4巴)。如通过激光衍射测量,干燥颗粒可以具有约1.5或更小的0.5/4巴分散性比(0.5/4)巴。干燥粉末可以具有总剂量为约25%或更多的小于5微米的fpf。本发明还涉及通过由吸入向有需要的受试者给予本文所述的干燥粉末来治疗真菌感染的方法。本发明还涉及通过由吸入向有需要的受试者给予本文所述的干燥粉末来治疗囊性纤维化的方法。本发明还涉及通过由吸入向有需要的受试者给予本文所述的干燥粉末来治疗哮喘的方法。本发明还涉及通过由吸入向有需要的受试者给予本文所述的干燥粉末来治疗曲霉菌病的方法。本发明还涉及通过由吸入向有需要的受试者给予本文所述的干燥粉末来治疗变应性支气管肺曲霉菌病(abpa)的方法。本发明还涉及通过由吸入向有需要的受试者给予本文所述的干燥粉末来治疗或降低呼吸系统疾病急性恶化的严重性的方法。本发明还涉及本文所述的干燥粉末用于治疗真菌感染。本发明还涉及本文所述的干燥粉末用于治疗曲霉菌病。本发明还涉及本文所述的干燥粉末用于治疗变应性支气管肺曲霉菌病(abpa)。本发明还涉及本文所述的干燥粉末用于治疗个体呼吸系统疾病的急性恶化。干燥粉末可以用基于胶囊的被动式干燥粉末吸入器递送到患者。本发明还涉及通过以下方法产生的干燥粉末:包括用任选的赋形剂喷雾干燥表面活性剂稳定的悬浮液,其中产生组成均匀的干燥颗粒。附图说明图1:配制品i和ii的颗粒x射线衍射图。图2:配制品iii和iv的颗粒x射线衍射图。图3:配制品v和vi的颗粒x射线衍射图。图4:配制品vii和viii的颗粒x射线衍射图。图5:配制品xi的颗粒x射线衍射图。图6:配制品xii的颗粒x射线衍射图。图7:配制品xiii的颗粒x射线衍射图。图8:配制品xiv的颗粒x射线衍射图。图9:配制品xv的颗粒x射线衍射图。图10:配制品xvi的颗粒x射线衍射图。图11:配制品xvii的颗粒x射线衍射图。图12:配制品xviii的颗粒x射线衍射图。图13:配制品xix的颗粒x射线衍射图。图14:在unidose中以60l/min从rs01雾化伊曲康唑粉末配制品后收集并且然后在usp装置ii设置中进行pod溶解测试的ism的累积质量溶解。图15:在unidose中以60l/min从rs01雾化伊曲康唑粉末配制品后收集并且然后在usp装置ii设置中进行pod溶解测试的ism的累积百分比质量溶解。图16a:不同粉末配制品中伊曲康唑溶解半衰期与颗粒大小之间的关系。图16b:不同粉末配制品中伊曲康唑溶解半衰期与表面积之间的关系。图17:不同粉末配制品中伊曲康唑溶解半衰期与cmax之间的关系。图18:表示为与配制品xix比率的伊曲康唑不同粉末配制品的溶解半衰期与剂量调节的cmax之间的关系。图19:在unidose中以15l/min从micromist雾化器雾化伊曲康唑悬浮液配制品(配制品xx和xxi)后收集并且然后在usp装置ii设置中进行pod溶解测试的ism的累积百分比质量溶解。图20:来自沉积在cngi的第4阶段上的伊曲康唑不同粉末配制品的回收剂量的累积质量百分比。图21:不同粉末配制品中伊曲康唑溶解半衰期与cmax之间的关系。图22:表示为与配制品xix比率的伊曲康唑不同粉末配制品的扩散速率与剂量调节的cmax之间的关系。图23:来自沉积在cngi的第4阶段上的伊曲康唑雾化悬浮液配制品(配制品xx和xxi)的回收剂量的累积质量百分比。具体实施方式本披露内容涉及含有呈结晶微粒形式的抗真菌剂的可吸入干燥粉末。本发明人发现,含有呈无定形形式的抗真菌剂(如伊曲康唑)的干燥粉末配制品在以治疗剂量吸入时具有较短的肺停留时间、减小的肺对血浆的暴露率和对肺组织的不希望毒性作用。不希望受任何特定理论的束缚,据信该物质的结晶形式(例如,纳米结晶形式)在肺中具有较慢的溶解速率,从而在给予后24小时内提供更持续的暴露并且使全身暴露最小化。此外,在总剂量或暴露持续时间方面,在没有无定形给药的肺组织中观察到的局部毒性与肺组织对药物的总暴露无关。伊曲康唑对人或动物肺细胞没有已知活性,并且因此增加局部浓度没有局部药理活性来解释局部毒性。相反,无定形形式的毒性似乎与仅次于伊曲康唑的无定形性质的溶解度增加有关,从而导致该药物在间质空间中过饱和,并且该组织中产生的重结晶导致局部、肉芽肿性炎症。令人惊讶的是,本发明人发现含有呈结晶微粒形式的抗真菌剂的干燥粉末对肺组织的毒性较小。这是令人惊讶的,因为结晶微粒抗真菌剂与无定形形式相比具有较低的溶解速率,并且比相应剂量呈无定形形式的抗真菌剂在肺中保留更长时间。抗真菌剂的结晶度以及抗真菌结晶颗粒的大小似乎对于有效治疗和减少肺部毒性是重要的。不希望受任何特定理论的束缚,据信抗真菌剂的结晶颗粒将比较大结晶颗粒更快速地溶解在气道内液中-部分是由于较大的表面积总量。还据信结晶抗真菌剂将比无定形抗真菌剂更慢地溶解在气道内液中。因此,本文所述的干燥粉末可以使用呈结晶微粒形式的抗真菌剂配制,其提供所希望的结晶度和颗粒大小,并且可以被定制以实现所希望的药代动力学特性,同时避免肺中不可接受的毒性。本披露内容的可吸入干燥粉末包括均匀的可吸入干燥颗粒,这些均匀的可吸入干燥颗粒含有1)呈结晶微粒形式的抗真菌剂,2)稳定剂,和任选的3)一种或多种赋形剂。因此,干燥粉末的特征在于可吸入干燥颗粒,这些可吸入干燥颗粒含有稳定剂、任选的一种或多种赋形剂、和含有结晶抗真菌剂的亚颗粒(小于可吸入干燥颗粒的颗粒)。此类可吸入干燥颗粒可以使用任何适合的方法制备,如通过制备其中呈结晶微粒形式的抗真菌剂悬浮在赋形剂水性溶液中的原料,并喷雾干燥该原料。干燥粉末可以通过吸入(如口腔吸入)向患者给予。为了实现口腔吸入,可以使用干燥粉末吸入器,如被动式干燥粉末吸入器。干燥粉末配制品可以用于治疗或预防患者的真菌感染,如曲霉菌感染。将受益于干燥粉末的患者是例如患有囊性纤维化、哮喘、和/或由于严重免疫功能低下而处于发展真菌感染风险中的那些患者。抗真菌剂(例如伊曲康唑)的吸入配制品在治疗这些患者时使口服或静脉内(iv)配制品的许多缺点最小化。定义如本文所用,术语“约”是指所述值的加或减5%的相对范围,例如,“约20mg”将是“20mg加或减1mg”。如本文所用,可吸入干燥颗粒的术语“给予(administration)”或“给予(administering)”是指将可吸入干燥颗粒引入受试者的呼吸道。如本文所用,术语“无定形”表示当通过粉末x射线衍射(xrd)分析时缺乏显著的结晶度。如本文所用的术语“胶囊发射的粉末质量”或“cepm”是指在吸入操作期间从胶囊或剂量单位容器发射的干燥粉末配制品的量。典型地通过在吸入操作之前和之后称重胶囊来重量测量cepm,以确定移除的粉末配制品的质量。cepm可以表示为移除的粉末质量(以毫克计),或者吸入操作之前胶囊中初始填充的粉末质量的百分比。如本文所用的术语“结晶微粒形式”是指抗真菌剂(包括其药学上可接受的形式,包括盐、水合物、对映体等),该抗真菌剂呈颗粒(即,小于包含本文所披露干燥粉末的可吸入干燥颗粒的亚颗粒)形式,并且其中抗真菌剂是至少约50%结晶。抗真菌剂的结晶度百分比是指呈结晶形式的化合物相对于亚颗粒中存在的化合物总量的百分比。如果希望,则抗真菌剂可以是至少约60%、至少约70%、至少约80%、至少约90%、至少约95%、或约100%结晶。呈结晶微粒形式的抗真菌剂呈体积中值直径为约50纳米(nm)至约5,000nm(dv50),优选80nm至1750nmdv50、或优选50nm至800nmdv50的颗粒形式。术语“可分散的”是描述干燥粉末或可吸入干燥颗粒被驱散成可吸入气溶胶的特征的术语。在一个方面,干燥粉末或可吸入干燥颗粒的分散性在本文中表示为如使用helos/rodos通过激光衍射测量,在1巴的分散(即调节器)压力下测量的体积中值几何直径(vmgd)除以在4巴的分散(即调节器)压力下测量的vmgd的商,或0.5巴下的vmgd除以4巴下的vmgd的商。这些商在本文中分别称为“1巴/4巴分散性比”和“0.5巴/4巴分散性比”,并且分散性与低的商相关。例如,1巴/4巴分散性比是指如通过helos或其他激光衍射系统测量的在约1巴下从rodos干燥粉末分散器(或等同技术)的孔发射的干燥粉末或可吸入干燥颗粒的vmgd,除以通过helos/rodos在4巴下测量的相同干燥粉末或可吸入干燥颗粒的vmgd。因此,高度可分散的干燥粉末或可吸入干燥颗粒将具有接近1.0的1巴/4巴分散性比或0.5巴/4巴分散性比。高度可分散的粉末具有低团聚、聚集或结块在一起的倾向和/或如果团聚、聚集或结块在一起,则当它们从吸入器发射并由受试者吸入时容易分散或解团聚。在另一方面,通过测量作为流速函数的从吸入器发射的颗粒大小来评估分散性。随着通过吸入器的流速降低,可用于转移到粉末以使其分散的气流中的能量减少。高度可分散的粉末将具有如通过其质量中值空气动力学直径(mmad)在空气动力学上表征或通过其vmgd在几何上表征的大小分布,其在由人吸入的典型流速范围(如约15至约60升/每分钟(lpm)、约20至约60lpm、或约30lpm至约60lpm)内基本上不增加。即使在较低的吸入流速下,高度可分散的粉末也将具有约80%或更大的发射的粉末质量或剂量、或胶囊发射的粉末质量或剂量。vmgd还可以称为体积中值直径(vmd)、x50、或dv50。如本文所用的术语“干燥颗粒”是指可以含有总计至多约15%水和/或另一种溶剂的可吸入颗粒。优选地,按干燥颗粒的重量计,干燥颗粒含有总计至多约10%、总计至多约5%、总计至多约1%、或总计0.01%与1%之间的水和/或另一种溶剂,或者可以基本上不含水和/或其他溶剂。如本文所用的术语“干燥粉末”是指包含可吸入干燥颗粒的组合物。干燥粉末可以含有总计至多约15%的水和/或另一种溶剂。优选地,按干燥粉末的重量计,干燥粉末含有总计至多约10%、总计至多约5%、总计至多约1%、或总计0.01%与1%之间的水和/或另一种溶剂,或者可以基本上不含水和/或其他溶剂。在一个方面,干燥粉末是可吸入干燥粉末。如本文所用,术语“有效量”是指实现所希望效果所需的药剂量;如在患者(例如囊性纤维化(cf)患者、哮喘患者和免疫功能低下的患者)的呼吸道中治疗真菌感染,例如曲霉菌感染;治疗变应性支气管肺曲霉菌病(abpa);以及治疗或降低呼吸系统疾病急性恶化的发病率或严重性。特定用途的实际有效量可以根据具体的干燥粉末或可吸入干燥颗粒、给予方式,和受试者的年龄、体重、一般健康状况,以及所治疗的症状或病状的严重性而变化。有待给予的干燥粉末和干燥颗粒的适合量以及特定患者的剂量时间表可以由普通技术临床医生基于这些和其他考虑因素来确定。如本文所用,术语“发射剂量”或“ed”是指在烧制或分散事件后从适合的吸入器装置递送药物配制品的指示。更具体地,对于干燥粉末配制品,ed是从单位剂量包装抽出并离开吸入器装置的接口管的粉末百分比的量度。ed被定义为由吸入器装置递送的剂量与标称剂量(即,在烧制前置于适合的吸入器装置中的每单位剂量的粉末质量)的比率。ed是实验测量的参数,并且可以使用unitedstatespharmacopeiaconvention[美国药典公约],罗克维尔市,马里兰州,第13版,222-225,2007的usp第601节气溶胶,计量剂量吸入器和干燥粉末吸入器,递送剂量均匀性,从干燥粉末吸入器取样递送剂量的方法测定。该方法利用体外装置设置以模拟患者给药。如本文所用的术语“标称剂量”是指大于或等于1mg抗真菌剂的个体剂量。标称剂量是用一个胶囊、泡罩、或安瓿吸入/给予的总剂量。如本文所用的术语“fpf(<x)”、“fpf(<x微米)”和“小于x微米的细颗粒分数”(其中x等于,例如3.4微米、4.4微米、5.0微米或5.6微米)是指具有小于x微米的空气动力学直径的干燥颗粒样品的分数。例如,fpf(<x)可以通过以下测定:将沉积在第二阶段和两阶段坍塌andersen级联冲击器(aci)的最终收集过滤器上的可吸入干燥颗粒的质量除以称入胶囊中以用于递送到仪器的可吸入干燥颗粒的质量。该参数还可以标识为“fpf_td(<x)”,其中td意指总剂量。可以使用八阶段aci进行类似的测量。标准60l/min流速下的八阶段aci截止值是不同的,但fpf_td(<x)可以从八阶段完整数据集推断出。八阶段aci结果还可以通过usp方法计算,该方法使用在aci中收集的剂量而不是胶囊中的剂量来确定fpf。类似地,可以使用七阶段下一代冲击器(ngi)。如本文所用的术语“fpd(<x)”、“fpd<x微米”、“fpd(<x微米)”和“小于x微米的细颗粒剂量”(其中x等于,例如3.4微米、4.4微米、5.0微米或5.6微米)是指由具有小于x微米的空气动力学直径的可吸入干燥颗粒递送的治疗剂的质量。fpd<x微米可以通过以下测定:在标准60l/min流速下使用八阶段aci并将沉积在最终收集过滤器上的质量求和,并直接计算或外推fpd值。如本文所用的术语“可吸入”是指干燥颗粒或干燥粉末,其适于通过吸入递送到受试者的呼吸道(例如,肺部递送)。可吸入干燥粉末或干燥颗粒具有小于约10微米、优选约5微米或更小的质量中值空气动力学直径(mmad)。如本文所用,术语“呼吸道”包括上呼吸道(例如鼻腔通道、鼻腔、喉咙、和咽部)、呼吸气道(例如喉、气管、支气管、和细支气管)和肺部(例如呼吸细支气管、肺泡管、肺泡囊、和肺泡)。本文中用于描述可吸入干燥颗粒的术语“小”是指具有约10微米或更小、优选约5微米或更小、或小于5微米的体积中值几何直径(vmgd)的颗粒。如本文所用的术语“稳定剂”是指当呈结晶微粒形式的抗真菌剂悬浮于其难以溶解在其中的液体中时改善抗真菌剂物理稳定性的化合物(例如,减少微粒的聚集、团聚、奥斯特瓦尔德成熟和/或絮凝)。适合的稳定剂是表面活性剂和两亲物质,并且包括聚山梨醇酯(ps;聚氧乙烯化脱水山梨糖醇脂肪酸酯),如ps20、ps40、ps60和ps80;脂肪酸,如月桂酸、棕榈酸、肉豆蔻酸、油酸和硬脂酸;脱水山梨糖醇脂肪酸酯,如span20、snap40、span60、span80、和span85;磷脂,如二棕榈酰磷脂酰胆碱(dppc)、1,2-二棕榈酰-sn-甘油-3-磷酸-l-丝氨酸(dpps)、1,2-二棕榈酰-sn-甘油-3-磷酸胆碱(dspc)、1-棕榈酰-2-油酰磷脂酰胆碱(popc)、和1,2-二油酰基-sn-甘油-3-磷酸胆碱(dopc);磷脂酰甘油(pg),如二磷脂酰甘油(dppg)、dspg、dppg、popg等;1,2-二硬脂酰基-sn-甘油-3-磷酸乙醇胺(dspe);脂肪醇;苄醇、聚氧乙烯-9-月桂基醚;甘氨胆酸盐;枯草菌表面活性剂;泊洛沙姆;聚乙烯吡咯烷酮(pvp);peg/ppg嵌段共聚物(普朗尼克/泊洛沙姆);聚氧乙烯胆甾醇醚;poe烷基醚;四丁酚醛;卵磷脂;等。优选的稳定剂是聚山梨醇酯和脂肪酸。特别优选的稳定剂是ps80。另一种优选的稳定剂是油酸。如本文所用的术语“均匀的干燥颗粒”是指含有作为表面活性剂稳定的悬浮液预处理的结晶药物(例如纳米结晶药物)的颗粒。则均匀的干燥颗粒通过以下形成:用(任选的)赋形剂喷雾干燥表面活性剂稳定的悬浮液,从而产生干燥颗粒,其组成均匀,或更具体地,其表面活性剂涂覆的结晶药物颗粒和任选一种或多种赋形剂的组成相同。干燥粉末和干燥颗粒本发明涉及包含可吸入干燥颗粒的干燥粉末配制品,这些可吸入干燥颗粒含有1)呈结晶微粒形式的抗真菌剂,2)稳定剂,和3)一种或多种赋形剂。任何希望的抗真菌剂可以包括在本文所述的配制品中。许多抗真菌剂是熟知的,例如,多烯抗真菌剂,如两性霉素b;三唑抗真菌剂,如伊曲康唑、酮康唑、氟康唑、伏立康唑、和泊沙康唑;棘白菌素抗真菌剂,如卡泊芬净、米卡芬净、和阿尼芬净。其他三唑抗真菌剂包括克霉唑、艾沙康唑、和咪康唑。包括一类新的化学类别的三萜系葡聚糖合成酶抑制剂,例如scy-078。还包括orotomide抗真菌剂,如f901318,其抑制二氢乳清酸脱氢酶。抗真菌剂的结晶度以及抗真菌亚颗粒的大小似乎对于有效治疗和减少肺部毒性是重要的。不希望受任何特定理论的束缚,据信呈结晶形式的抗真菌剂的较小亚颗粒将比较大颗粒更快速地溶解在气道内液中-部分是由于较大的表面积量。还据信结晶抗真菌剂将比无定形抗真菌剂更慢地溶解在气道内液中。因此,本文所述的干燥粉末可以使用呈结晶微粒形式的抗真菌剂配制,其提供所希望的结晶度和亚颗粒大小,并且可以被定制以实现所希望的药代动力学特性,同时避免肺中不可接受的毒性。可吸入干燥颗粒含有按重量计(wt%)约1%至约95%的抗真菌剂。优选的是可吸入干燥颗粒含有一定量的抗真菌剂,使得可以给予和维持治疗有效剂量,而不需要每天吸入大量干燥粉末多于三次。例如,优选的是可吸入干燥颗粒含有按重量计(wt%)约10%至75%、约15%至75%、约25%至75%、约30%至70%、约40%至60%、约20%、约50%、或约70%的抗真菌剂。可吸入干燥颗粒可以含有按重量计(wt%)约75%、约80%、约85%、约90%、或约95%的抗真菌剂。在特定实施例中,可吸入干燥颗粒中的抗真菌剂的范围是按重量计(wt%)约40%至约90%、约55%至约85%、约55%至约75%、或约65%至约85%。按重量计存在于可吸入干燥颗粒中的抗真菌剂的量还称为“药物负载”。抗真菌剂以结晶微粒形式(例如,纳米结晶)存在于可吸入干燥颗粒中。更具体地,呈约50nm至约5,000nm(dv50)的亚颗粒形式,优选地,其中抗真菌剂是至少50%结晶。例如,对于任何希望的药物负载,亚颗粒大小可以是约100nm、约300nm、约1500nm、约80nm至约300nm、约80nm至约250nm、约80nm至约200nm、约100nm至约150nm、约1200nm至约1500nm、约1500nm至约1750nm、约1200nm至约1400nm、或约1200nm至约1350nm(dv50)。在特定实施例中,亚颗粒是约50nm至约2500nm之间、约50nm与1000nm之间、约50nm与800nm之间、约50nm与600nm之间、约50nm与500nm之间、约50nm与400nm之间、约50nm与300nm之间、约50nm与200nm之间、或约100nm与300nm之间。此外,对于任何希望的药物负载和亚颗粒大小,抗真菌剂的结晶度可以是至少约50%、至少约60%、至少约70%、至少约80%、至少约90%、至少约95%、或约100%结晶。优选地,抗真菌剂是约100%结晶。呈结晶微粒形式的抗真菌剂可以使用适合的方法,包括稳定剂(如果希望),如通过湿磨、喷射研磨或其他适合的方法制备成任何希望的亚颗粒大小。可吸入干燥颗粒还包括稳定剂。稳定剂有助于在湿磨过程中,在喷雾干燥原料中保持呈结晶微粒形式的抗真菌剂的所希望大小,并有助于润湿和分散。优选使用如所需的尽可能少的稳定剂以获得希望的干燥粉末。稳定剂的量典型地与存在于干燥颗粒中的抗真菌剂的量有关,并且可以在从约1:1(抗真菌剂:稳定剂(wt:wt))至约50:1(wt:wt)范围内,其中≥(大于或等于)10:1是优选的。例如,干燥颗粒中抗真菌剂:稳定剂(wt:wt)的比率可以≥(大于或等于)10:1,约10:1、约20:1、或约10:1至约20:1。在特定实施例中,该比率是约5:1至约20:1、约7:1至约15:1、或约9:1至约11:1。此外,存在于干燥颗粒中的稳定剂的量可以在按重量计(wt%)约0.05%至约45%范围内。在特定实施例中,该范围是按重量计(wt%)约1%至约15%、约4%至约10%、或约5%至约8%。通常优选的是可吸入干燥颗粒含有按重量计(wt%)少于约10%的稳定剂,如7wt%、5wt%或1wt%。可替代地,可吸入干燥颗粒含有约5wt%、约6wt%、约7wt%、约7.5wt%、约8wt%、或约10%稳定剂。特别优选的是可吸入干燥颗粒含有少于约5wt%的稳定剂。用于本文所述干燥粉末的特别优选的稳定剂是聚山梨醇酯80。另一种优选的稳定剂是油酸(或其盐形式)。与使用表面活性剂防止产生的干燥粉末中结晶开始的现有技术相反,加入本发明的表面活性剂以稳定结晶药物在抗溶剂中的胶体悬浮液。可吸入干燥颗粒还包括任何适合和希望量的一种或多种赋形剂。干燥颗粒可以含有约10wt%至约99wt%的总赋形剂含量,其中约25wt%至约85wt%、或约40wt%至约55wt%是更典型的。干燥颗粒可以含有约1wt%、约2wt%、约4wt%、约6wt%、约8wt%、或少于约10wt%的总赋形剂含量。在特定实施例中,该范围是约5%至约50%、约15%至约50%、约25%至约50%、约5%至约40%、约5%至约30%、约5%至约20%、或约5%至约15%。许多赋形剂是本领域熟知的并且可以包括在本文所述的干燥粉末和干燥颗粒中。对本文所述的干燥粉末和干燥颗粒特别优选的药学上可接受的赋形剂包括一价和二价金属阳离子盐、碳水化合物、糖醇和氨基酸。适合的一价金属阳离子盐,包括例如钠盐和钾盐。可以存在于本发明的可吸入干燥颗粒中的适合钠盐包括例如,氯化钠、柠檬酸钠、硫酸钠、乳酸钠、乙酸钠、碳酸氢钠、碳酸钠、硬脂酸钠、抗坏血酸钠、苯甲酸钠、二磷酸钠、磷酸钠、亚硫酸氢钠、硼酸钠、葡萄糖酸钠、偏硅酸钠等。适合的钾盐包括例如,氯化钾、溴化钾、碘化钾、碳酸氢钾、亚硝酸钾、过硫酸钾、亚硫酸钾、亚硫酸氢钾、磷酸钾、乙酸钾、柠檬酸钾、谷氨酸钾、鸟苷酸二钾、葡萄糖酸钾、苹果酸钾、抗坏血酸钾、山梨酸钾、琥珀酸钾、酒石酸钾钠及其任何组合。适合的二价金属阳离子盐,包括镁盐和钙盐。适合的镁盐包括例如,乳酸镁、氟化镁、氯化镁、溴化镁、碘化镁、磷酸镁、硫酸镁、亚硫酸镁、碳酸镁、氧化镁、硝酸镁、硼酸镁、乙酸镁、柠檬酸镁、葡萄糖酸镁、马来酸镁、琥珀酸镁、苹果酸镁、牛磺酸镁、乳清酸镁、甘氨酸镁、环烷酸镁、乙酰丙酮镁、甲酸镁、氢氧化镁、硬脂酸镁、六氟硅酸镁、水杨酸镁或其任何组合。适合的钙盐包括例如,氯化钙、硫酸钙、乳酸钙、柠檬酸钙、碳酸钙、乙酸钙、磷酸钙、海藻酸钙、硬脂酸钙、山梨酸钙、葡萄糖酸钙等。优选的钠盐是硫酸钠。优选的钠盐是氯化钠。优选的钠盐是柠檬酸钠。优选的镁盐是乳酸镁。在这方面有用的碳水化合物赋形剂包括单糖和多糖。代表性的单糖包括右旋糖(无水和一水合物;也称为葡萄糖和葡萄糖一水合物)、半乳糖、d-甘露糖、山梨糖等。代表性的二糖包括乳糖、麦芽糖、蔗糖、海藻糖等。代表性的三糖包括棉子糖等。如所希望的,可以使用其他碳水化合物赋形剂,包括葡聚糖、麦芽糖糊精和环糊精,如2-羟丙基-β-环糊精。代表性的糖醇包括甘露醇、山梨糖醇等。优选的糖醇是甘露醇。适合的氨基酸赋形剂包括以下中的任一种:在标准制药加工技术下形成粉末的天然存在的氨基酸,并且包括非极性(疏水)氨基酸和极性(不带电荷、带正电荷和带负电荷)的氨基酸,此类氨基酸是药品等级的并且通常被美国食品和药物管理局认为是安全的(gras)。非极性氨基酸的代表性实例包括丙氨酸、异亮氨酸、亮氨酸、蛋氨酸、苯丙氨酸、脯氨酸、色氨酸和缬氨酸。极性不带电荷的氨基酸的代表性实例包括胱氨酸、甘氨酸、谷氨酰胺、丝氨酸、苏氨酸、和酪氨酸。极性带正电荷的氨基酸的代表性实例包括精氨酸、组氨酸和赖氨酸。带负电荷的氨基酸的代表性实例包括天冬氨酸和谷氨酸。优选的氨基酸是亮氨酸。本文所述的干燥颗粒含有1)呈结晶微粒形式的抗真菌剂,2)稳定剂,和任选的3)一种或多种赋形剂。在一些方面,干燥颗粒含有第一赋形剂,其为一价或二价金属阳离子盐,和第二赋形剂,其为氨基酸、碳水化合物或糖醇。例如,第一赋形剂可以是钠盐或镁盐,并且第二赋形剂可以是氨基酸(如亮氨酸)。在更具体的实例中,第一赋形剂可以是硫酸钠、氯化钠或乳酸镁,并且第二赋形剂可以是亮氨酸。甚至更具体地,第一赋形剂可以是硫酸钠并且第二赋形剂可以是亮氨酸。在另一个实例中,第一赋形剂可以是钠盐或镁盐,并且第二赋形剂可以是糖醇(如甘露醇)。在更具体的实例中,第一赋形剂可以是硫酸钠、氯化钠或乳酸镁,并且第二赋形剂可以是甘露醇。在其他实例中,干燥颗粒包括呈结晶微粒形式的抗真菌剂、稳定剂和一种赋形剂,例如钠盐、镁盐或氨基酸(例如亮氨酸)。在一个方面,本发明涉及包含可吸入干燥颗粒的干燥粉末配制品,这些可吸入干燥颗粒包含1)呈结晶微粒形式的抗真菌剂,2)稳定剂,和3)一种或多种赋形剂,其条件是抗真菌剂不是多烯抗真菌剂(例如,两性霉素b)。在一个优选的方面,本发明涉及包含可吸入干燥颗粒的干燥粉末配制品,这些可吸入干燥颗粒包含1)呈结晶微粒形式的三唑抗真菌剂,2)稳定剂,和3)一种或多种赋形剂。在一个方面,本发明涉及包含可吸入干燥颗粒的干燥粉末配制品,这些可吸入干燥颗包含约50%至约80%的呈结晶微粒形式的三唑抗真菌剂、约4%至约40%的稳定剂,和约1%至约9%的一种或多种赋形剂;约45%至约85%的呈结晶微粒形式的三唑抗真菌剂、约3%至约15%的稳定剂、约3%至约40%的钠盐,和约1%至约9%的一种或多种氨基酸;约45%至约85%的呈结晶微粒形式的三唑抗真菌剂、约3%至约15%的稳定剂、约3%至约40%的硫酸钠,和约1%至约9%的亮氨酸;其中所有百分比均为重量百分比,并且所有配制品在干基的基础上加起来为100%。在特别优选的方面,本发明涉及包含可吸入干燥颗粒的干燥粉末配制品,这些可吸入干燥颗粒包含1)呈结晶微粒形式的伊曲康唑,2)稳定剂,和3)一种或多种赋形剂。在该特别优选的方面,干燥粉末配制品不包含乳糖。在一个方面,本发明涉及一种干燥粉末配制品,该干燥粉末配制品包含50%伊曲康唑、35%硫酸钠、10%亮氨酸、和5%聚山梨醇酯80。在一个方面,本发明涉及一种干燥粉末配制品,该干燥粉末配制品包含50%伊曲康唑、37%硫酸钠、8%亮氨酸、和5%聚山梨醇酯80。在另一方面,本发明涉及一种干燥粉末配制品,该干燥粉末配制品包含60%伊曲康唑、26%硫酸钠、8%亮氨酸、和6%聚山梨醇酯80。在另一方面,本发明涉及一种干燥粉末配制品,该干燥粉末配制品包含70%伊曲康唑、15%硫酸钠、8%亮氨酸、和7%聚山梨醇酯80。在另一方面,本发明涉及一种干燥粉末配制品,该干燥粉末配制品包含75%伊曲康唑、9.5%硫酸钠、8%亮氨酸、和7.5%聚山梨醇酯80。在另一方面,本发明涉及一种干燥粉末配制品,该干燥粉末配制品包含80%伊曲康唑、4%硫酸钠、8%亮氨酸、和8%聚山梨醇酯80。在另一方面,本发明涉及一种干燥粉末配制品,该干燥粉末配制品包含80%伊曲康唑、10%硫酸钠、2%亮氨酸、和8%聚山梨醇酯80。在另一方面,本发明涉及一种干燥粉末配制品,该干燥粉末配制品包含80%伊曲康唑、11%硫酸钠、1%亮氨酸、和8%聚山梨醇酯80。干燥粉末和/或可吸入干燥颗粒优选地是小的、质量密集的、和可分散的。为了测量体积中值几何直径(vmgd),可以使用激光衍射系统,例如spraytec系统(颗粒大小分析仪器,马尔文仪器公司(malverninstruments))和helos/rodos系统(具有干式分配单元的激光衍射传感器,德国新帕泰克有限公司(sympatecgmbh))。如使用helos/rodos系统在最大孔环压力下以1.0巴的分散压力设定(也称为调节器压力)通过激光衍射测量,可吸入干燥颗粒具有约10微米或更小、约5微米或更小、约4μm或更小、约3μm或更小、约1μm至约5μm、约1μm至约4μm、约1.5μm至约3.5μm、约2μm至约5μm、约2μm至约4μm、或约2μm至约3μm的vmgd。优选地,vmgd是约5微米或更小或约4μm或更小。在一个方面,干燥粉末和/或可吸入干燥颗粒具有约0.5微米或约1.0微米的最小vmgd。干燥粉末和/或可吸入干燥颗粒优选地具有小于约2.0(例如约0.9至小于约2)、约1.7或更小(例如约0.9至约1.7)、约1.5或更小(例如约0.9至约1.5)、约1.4或更小(例如约0.9至约1.4)、或约1.3或更小(例如约0.9至约1.3)的1巴/4巴分散性比和/或0.5巴/4巴分散性比,并且优选地具有约1.5或更小(例如约1.0至约1.5)、和/或约1.4或更小(例如约1.0至约1.4)的1巴/4巴和/或0.5巴/4巴。干燥粉末和/或可吸入干燥颗粒优选地具有至少约0.2g/cm3、至少约0.25g/cm3的振实密度,至少约0.3g/cm3、至少约0.35g/cm3的振实密度,至少0.4g/cm3的振实密度。例如,干燥粉末和/或可吸入干燥颗粒具有大于0.4g/cm3(例如,大于0.4g/cm3至约1.2g/cm3)的振实密度,至少约0.45g/cm3(例如,约0.45g/cm3至约1.2g/cm3)、至少约0.5g/cm3(例如,约0.5g/cm3至约1.2g/cm3)、至少约0.55g/cm3(例如,约0.55g/cm3至约1.2g/cm3)、至少约0.6g/cm3(例如,约0.6g/cm3至约1.2g/cm3)或至少约0.6g/cm3至约1.0g/cm3的振实密度。可替代地,干燥粉末和/或可吸入干燥颗粒优选地具有约0.01g/cm3至约0.5g/cm3、约0.05g/cm3至约0.5g/cm3、约0.1g/cm3至约0.5g/cm3、约0.1g/cm3至约0.4g/cm3、或约0.1g/cm3至约0.4g/cm3的振实密度。可替代地,干燥粉末和/或可吸入干燥颗粒具有约0.15g/cm3至约1.0g/cm3的振实密度。干燥粉末和/或可吸入干燥颗粒具有至少约0.1g/cm3或至少约0.8g/cm3的本体密度。例如,干燥粉末和/或可吸入干燥颗粒具有约0.1g/cm3至约0.6g/cm3、约0.2g/cm3至约0.7g/cm3、约0.3g/cm3至约0.8g/cm3的本体密度。当干燥粉末是可吸入干燥粉末时,可吸入干燥颗粒和干燥粉末优选地具有小于10微米的mmad,优选约5微米或更小、或约4微米或更小的mmad。在一个方面,可吸入干燥粉末和/或可吸入干燥颗粒优选地具有约0.5微米或约1.0微米的最小mmad。在一个方面,可吸入干燥粉末和/或可吸入干燥颗粒优选地具有约2.0微米、约3.0微米、或约4.0微米的最小mmad。干燥粉末和/或可吸入干燥颗粒优选地具有总剂量为至少约35%、优选至少约45%、至少约60%、约45%与约80%之间、或约60%与约80%之间的小于约5.6微米的fpf(fpf<5.6μm)。干燥粉末和/或可吸入干燥颗粒优选地具有总剂量为至少约20%、优选至少约25%、至少约30%、至少约40%、约25%与约60%之间、或约40%与约60%之间的小于约3.4微米的fpf(fpf<3.4μm)。干燥粉末和/或可吸入干燥颗粒优选地具有按重量计至多约15%、按重量计至多约10%、按重量计至多约5%、按重量计至多约1%、或约0.01%与约1%之间的总水和/或溶剂含量,或者可以基本上不含水或其他溶剂。干燥粉末和/或可吸入干燥颗粒优选地可以以低吸入能量给予。为了使不同吸入流速、体积的粉末的分散和不同阻力的吸入器相关,可以计算进行吸入操作所需的能量。吸入能量可以从方程e=r2q2v计算,其中e是以焦耳计的吸入能量,r是以kpa1/2/lpm计的吸入器阻力,q是以l/min计的稳定流速,并且v是以l计的吸入空气体积。基于fda关于干燥粉末吸入器的指导文件和tiddens等人(journalofaerosolmed[气溶胶医学杂志],19(4),第456-465页,2006)的工作(他们发现成人通过各种dpi的吸入体积平均为2.2l),通过使用由clarke等人(journalofaerosolmed[气溶胶医学杂志],6(2),第99-110页,1993)针对来自0.02和0.055kpa1/2/lpm的两个吸入器阻力的流速q(同时吸入体积为2l)测量的峰值吸入流速(pifr)值,预测健康成人群体能够实现范围从舒适吸入的2.9焦耳至最大吸入的22焦耳的吸入能量。预测轻度、中度和重度成人copd患者能够分别实现5.1至21焦耳、5.2至19焦耳、和2.3至18焦耳的最大吸入能量。这再次基于使用测量的pifr值来计算吸入能量方程中的流速q。每组可实现的pifr是吸入通过的吸入器阻力的函数。将broeders等人的工作(eurrespirj[欧洲呼吸杂志],18,第780-783页,2001)用于预测通过两个电阻各自为0.021和0.032kpa1/2/lpm的干燥粉末吸入器可实现的最大和最小pifr。类似地,基于与copd群体相同的假设和来自broeders等人的pifr数据,预测成人哮喘患者能够实现7.4至21焦耳的最大吸入能量。例如,健康的成人和儿童、copd患者、5岁及以上的哮喘患者、和cf患者能够提供足够的吸入能量来清空和分散本发明的干燥粉末配制品。干燥粉末和/或可吸入干燥颗粒优选地特征在于高发射剂量,如从被动式干燥粉末吸入器受试者的至少75%、至少80%、至少85%、至少90%、至少95%的cepm至约5焦耳、约3.5焦耳、约2.4焦耳、约2焦耳、约1焦耳、约0.8焦耳、约0.5焦耳、或约0.3焦耳的总吸入能量被应用于干燥粉末吸入器。保持干燥粉末和/或可吸入干燥颗粒的容器可以含有约5mg、约7.5mg、约10mg、约15mg、约20mg、或约30mg。在一个方面,干燥粉末和/或可吸入干燥颗粒的特征在于当在以下条件下从具有约0.036sqrt(kpa)/升/分钟的阻力的被动式干燥粉末吸入器发射时,cepm为80%或更大并且vmgd为5微米或更小:空气流速为30lpm,使用含有10mg总质量的3号胶囊运行3秒。在另一方面,干燥粉末和/或可吸入干燥颗粒的特征在于当在以下条件下从具有约0.036sqrt(kpa)/升/分钟的阻力的被动式干燥粉末吸入器发射时,cepm为80%或更大并且vmgd为5微米或更小:空气流速为20lpm,使用含有10mg总质量的3号胶囊运行3秒。在另一方面,干燥粉末和/或可吸入干燥颗粒的特征在于当在以下条件下从具有约0.036sqrt(kpa)/升/分钟的阻力的被动式干燥粉末吸入器发射时,cepm为80%或更大并且vmgd为5微米或更小:空气流速为15lpm,使用含有10mg总质量的4号胶囊运行3秒。干燥粉末可以填充单位剂量容器,或单位剂量容器可以至少2%满、至少5%满、至少10%满、至少20%满、至少30%满、至少40%满、至少50%满、至少60%满、至少70%满、至少80%满、或至少90%满。单位剂量容器可以是胶囊(例如,000、00、0e、0、1、2、3、和4号,各自的体积容量为1.37ml、950μl、770μl、680μl、480μl、360μl、270μl、和200μl)。该胶囊可以至少约2%满、至少约5%满、至少约10%满、至少约20%满、至少约30%满、至少约40%满、或至少约50%满。单位剂量容器可以是泡罩。泡罩可以包装为单个泡罩或一组泡罩(例如7个泡罩、14个泡罩、28个泡罩或30个泡罩)的一部分。一个或多个泡罩可以优选地至少30%满、至少50%满或至少70%满。本发明的一个优点是生产的粉末在宽流速范围内良好分散并且相对流动独立。本发明的干燥粉末和/或可吸入干燥颗粒使得能够为广泛的患者群体使用简单的被动式dpi。在特定方面,本发明涉及干燥粉末和/或可吸入干燥颗粒,其包含呈结晶微粒形式(例如,约80nm至约1750nm,如约60nm至约175nm、约150nm至约400nm或约1200nm至约1750nm的颗粒)的抗真菌剂、稳定剂、和任选的一种或多种赋形剂。特定的干燥粉末和可吸入干燥颗粒具有表1中所示的以下配制品。本文所述的干燥粉末和/或可吸入干燥颗粒优选地特征在于:1)如使用helos/rodos系统测量的约10微米或更小,优选约5微米或更小的1巴下的vmgd;2)约1.5或更小、约1.4或更小或约1.3或更小的1巴/4巴分散性比和/或0.5巴/4巴分散性比;3)约10微米或更小、优选约5微米或更小的mmad;4)总剂量为至少约45%或至少约60%的fpf<5.6μm;和/或5)总剂量为至少约25%或至少约40%的fpf<3.4μm。如果希望,则干燥粉末和/或可吸入干颗粒的特征进一步在于约0.2g/cm3或更大、约0.3g/cm3或更大、约0.4g/cm3或更大、大于0.4g/cm3、约0.45g/cm3或更大或约0.5g/cm3或更大的振实密度。由前一段落中表征的任何范围或具体披露的配制品描述的干燥粉末和/或可吸入干燥颗粒可以填充到容器(例如胶囊或泡罩)中。当容器是胶囊时,该胶囊例如是2号或3号胶囊,并且优选是3号胶囊。胶囊材料可以是例如明胶或hpmc(羟丙基甲基纤维素),并且优选地是hpmc。以上描述和表征的干燥粉末和/或可吸入干燥颗粒可以包含在干燥粉末吸入器(dpi)中。该dpi可以是基于胶囊的dpi或基于泡罩的dpi,并且优选基于胶囊的dpi。更优选地,干燥粉末吸入器选自rs01系列干燥粉末吸入器(意大利plastiapes.p.a公司)。更优选地,干燥粉末吸入器选自rs01hr或rs01uhr2。最优选地,干燥粉末吸入器是rs01hr。用于制备干燥粉末和干燥颗粒的方法可吸入干燥颗粒和干燥粉末可以使用任何适合的方法制备,其条件是干燥粉末配制品不能是即时分散体。用于制备干燥粉末和/或可吸入干燥颗粒的许多适合方法是本领域常规的,并且包括单乳液和双乳液溶剂蒸发、喷雾干燥、喷雾冷冻干燥、研磨(例如喷射研磨)、共混、溶剂萃取、溶剂蒸发、相分离、简单和复杂的凝聚、界面聚合、涉及使用超临界二氧化碳(co2)、超声波结晶、纳米颗粒聚集体形成的适合方法和其他适合方法(包括其组合)。可以使用本领域已知的用于制备微球或微胶囊的方法制备可吸入干燥颗粒。这些方法可以在导致形成具有所希望空气动力学特性(例如空气动力学直径和几何直径)的可吸入干燥颗粒的条件下采用。如果希望,则可以使用适合方法(如筛选)选择具有所希望特性(如大小和密度)的可吸入干燥颗粒。用于选择具有所希望特性(如大小和密度)的可吸入干燥颗粒的适合方法包括湿筛分、干筛分、和空气动力学分级器(如旋风分离器)。优选地喷雾干燥可吸入干燥颗粒。适合的喷雾干燥技术例如由k.masters描述于“spraydryinghandbook[喷雾干燥手册]”,johnwiley&sons[约翰威利出版社],纽约,1984中。一般来说,在喷雾干燥期间,来自热气体(如加热空气或氮气)的热量用于从通过雾化连续液体进料形成的液滴蒸发溶剂。当使用热空气时,空气中的水分在使用前被至少部分地除去。当使用氮气时,氮气可以“干燥”运行,这意味着没有另外的水蒸气与气体组合。如果希望,则可以在喷雾干燥运行开始之前以高于“干燥”氮气的固定值设定氮气或空气的水分含量。如果希望,则用于制备干燥颗粒的喷雾干燥或其他仪器(例如喷射研磨仪器)可以包括内联几何颗粒分析仪,其确定可吸入干燥颗粒在产生时的几何直径,和/或内联空气动力学颗粒分析仪,其确定可吸入干燥颗粒在产生时的空气动力学直径。对于喷雾干燥,通过雾化装置将含有有待在适合溶剂(例如水性溶剂、有机溶剂、水性-有机混合物或乳液)中产生的干燥颗粒组分的溶液、乳液或悬浮液分配到干燥容器。例如,可以使用喷嘴或旋转雾化器将溶液或悬浮液分配到干燥容器。喷嘴可以是双流体喷嘴,其可以是内部混合设置或外部混合设置。可替代地,可以使用具有4或24叶轮的旋转雾化器。可以配备有旋转雾化器和/或喷嘴的适合喷雾干燥器的实例包括二者均由gea尼罗公司(geaniro)(丹麦)制造的mobileminorspraydryer或modelpsd-1、büchib-290minispraydryer(瑞士弗拉维尔的步琪实验室技术服务有限公司(labortechnikag,flawil,switzerland))、proceptformatrixr&d喷雾干燥器(比利时泽尔扎特的procept公司(proceptnv,zelzate,belgium)),以及其他几种喷雾干燥器选项。实际喷雾干燥条件将部分地取决于喷雾干燥溶液或悬浮液的组成和材料流速。普通技术人员将能够基于有待喷雾干燥的溶液、乳液或悬浮液的组成,所希望的颗粒特性和其他因素来确定适当的条件。通常,喷雾干燥器的入口温度是约90℃至约300℃。喷雾干燥器出口温度将取决于如进料温度和被干燥材料的特性等因素而变化。一般来说,出口温度是约50℃至约150℃。如果希望,则所产生的可吸入干燥颗粒可以例如使用筛子通过体积大小分级,或者例如使用旋风分离器通过空气动力学大小分级,和/或使用本领域技术人员已知的技术根据密度进一步分离。为了制备本发明的可吸入干燥颗粒,一般来说,制备含有所希望干燥粉末(即原料)组分的乳液或悬浮液,并在适合的条件下喷雾干燥。优选地,原料中溶解或悬浮的固体浓度是至少约1g/l、至少约2g/l、至少约5g/l、至少约10g/l、至少约15g/l、至少约20g/l、至少约30g/l、至少约40g/l、至少约50g/l、至少约60g/l、至少约70g/l、至少约80g/l、至少约90g/l或至少约100g/l。可以通过在适合的溶剂中溶解、悬浮、或乳化适合的组分(例如盐、赋形剂、其他活性成分)制备单一溶液、悬浮液或乳液来提供原料。溶液、乳液或悬浮液可以使用任何适合的方法制备,如干燥和/或液体组分的本体混合或液体组分的静态混合以形成组合。例如,可以使用静态混合器组合亲水性组分(例如水性溶液)和疏水性组分(例如有机溶液)以形成组合。然后可以将该组合雾化以产生液滴,将其干燥以形成可吸入干燥颗粒。优选地,在组分在静态混合器中混合之后立即进行雾化步骤。可替代地,在本体混合溶液上进行雾化步骤。原料可以使用其中呈微粒形式的抗真菌剂具有低溶解度的任何溶剂(如有机溶剂、水性溶剂或其混合物)来制备。可以采用的适合有机溶剂包括但不限于醇,例如像乙醇、甲醇、丙醇、异丙醇、丁醇等。其他有机溶剂包括但不限于四氢呋喃(thf)、全氟化碳、二氯甲烷、氯仿、乙醚、乙酸乙酯、甲基叔丁基醚等。可以采用的共溶剂包括水性溶剂和有机溶剂,如但不限于如上所述的有机溶剂。水性溶剂包括水和缓冲溶液。优选的溶剂是水。各种方法(例如静态混合、本体混合)可以用于混合溶质和溶剂以制备原料,这是本领域已知的。如果希望,则可以使用其他适合的混合方法。例如,引起或促进混合的另外组分可以包括在原料中。例如,二氧化碳产生嘶嘶声或泡腾,并且因此可以用于促进溶质和溶剂的物理混合。原料或原料组分可以具有任何所希望的ph、粘度或其他特性。如果希望,则可以将ph缓冲剂加入溶剂或共溶剂或形成的混合物中。一般来说,混合物的ph在从约3至约8范围内。可以制造干燥粉末和/或可吸入干燥颗粒,并且然后例如通过借助于旋风分离器进行的过滤或离心将其分离,以提供具有预选大小分布的颗粒样品。例如,样品中大于约30%、大于约40%、大于约50%、大于约60%、大于约70%、大于约80%、或大于约90%的可吸入干燥颗粒可以具有在选定范围内的直径。一定百分比的可吸入干燥颗粒落入的选定范围可以是例如本文所述的任何大小范围,如约0.1至约3微米之间的vmgd。悬浮液可以是纳米悬浮液,类似于用于制备含有纳米结晶药物的干燥粉末的中间体。干燥粉末可以是包埋在基质材料(如硫酸钠和亮氨酸)中的药物。任选地,可以将干燥粉末喷雾干燥,使得干燥颗粒小、致密、并且可分散。干燥粉末可以仅由本文所述的可吸入干燥颗粒组成,而没有其他载体或赋形剂颗粒(称为“纯粉末”)。如果希望,则干燥粉末可以包含本文所述的可吸入干燥颗粒与其他载体或赋形剂颗粒(如大于10微米、20微米至500微米,并且优选25微米与250微米之间的乳糖载体颗粒)的共混物。在优选的实施例中,干燥粉末不含有载体颗粒。在一个方面,结晶药物颗粒包埋在包含赋形剂和/或稳定剂的基质中。干燥粉末可以包含均匀含量的可吸入干燥颗粒,其中每个颗粒含有结晶药物。因此,如本文所用,“均匀含量”意指每个可吸入颗粒均含有一定量的呈结晶微粒形式的抗真菌剂、稳定剂、和赋形剂。干燥粉末可以包含可吸入干燥颗粒,其中至少98%、至少99%、或基本上所有颗粒(按重量计)均含有抗真菌剂。干燥粉末可以包含结晶药物颗粒,这些结晶药物颗粒分布在整个包含一种或多种赋形剂的基质中。赋形剂可以包含任何数量的盐、糖、脂质、氨基酸、表面活性剂、聚合物、或适于药学用途的其他组分。优选的赋形剂包括硫酸钠和亮氨酸。典型地通过首先使用任何数量本领域技术人员熟悉的技术(例如,湿磨、喷射研磨)处理结晶药物以调节颗粒大小来制造干燥粉末。将结晶药物在抗溶剂中与稳定剂一起加工以形成悬浮液。优选的稳定剂包括聚山梨醇酯(tween)80和油酸。然后用一种或多种另外的赋形剂干燥喷雾结晶药物的稳定悬浮液。所得干燥颗粒包含分散在整个赋形剂基质中的结晶药物,其中每个干燥颗粒具有均匀的组成。在特定的实施例中,本发明的干燥粉末通过以结晶药物(例如伊曲康唑)开始制备,该结晶药物通常可在微晶大小范围内获得。使用本领域技术人员熟悉的许多技术中的任一种将微晶药物的颗粒大小减小为纳米结晶大小,包括但不限于高压均化、高剪切均化、喷射研磨、针磨、微流化、或湿磨(也称为球磨、珍珠磨或珠磨)。湿磨经常是优选的,因为它能够实现宽范围的颗粒大小分布,包括纳米(<1μm)大小域中的那些。在亚微米大小域中变得特别重要的是使用表面稳定组分,如表面活性剂(例如吐温80)。表面活性剂能够在研磨期间产生亚微米颗粒并形成物理稳定的悬浮液,因为它们隔离了在研磨期间产生的许多高能表面,从而防止聚集和沉降。因此,表面活性剂的存在对于喷雾干燥均匀的微颗粒是重要的,因为表面活性剂允许形成均匀且稳定的悬浮液,从而确保跨颗粒的组成均匀性。表面活性剂的使用允许形成微悬浮液或纳米悬浮液。使用表面活性剂,将纳米结晶药物(例如itz)颗粒悬浮在抗溶剂中的稳定胶体悬浮液中。药物的抗溶剂可以利用水,或水和其他可混溶溶剂(如醇或酮)的组合作为胶体悬浮液的连续反溶剂相。喷雾干燥原料可以通过将可溶性组分溶解在一种或多种所希望溶剂中,随后在混合时将表面活性剂稳定的结晶药物纳米悬浮液分散在所得原料中来制备,尽管该方法不限于该特定操作顺序。用于分析干燥粉末和/或可吸入干燥颗粒的方法见于以下例证部分。治疗用途和方法本发明的干燥粉末和/或可吸入干燥颗粒适于给予呼吸道,例如给予有需要的受试者用于治疗呼吸(例如肺)疾病,如囊性纤维化、哮喘,尤其是严重的哮喘和严重免疫功能低下的患者。该治疗特别适用于治疗曲霉菌感染。该治疗还可用于治疗对伊曲康唑敏感的真菌感染。本发明的另一方面是例如在患有肺病(如哮喘或囊性纤维化)的患者中治疗变应性支气管肺曲霉菌病(abpa)。在其他方面,本发明是一种治疗、降低发病率或严重性或预防由呼吸道中的真菌感染(如曲霉菌感染)引起的急性恶化的方法。在另一方面,本发明是一种治疗、降低发病率或严重性或预防由呼吸道中的真菌感染(如曲霉菌感染)引起的恶化的方法。在另一方面,本发明是一种例如在患有肺病(如哮喘或囊性纤维化)的患者中治疗、降低发病率或严重性或预防由变应性支气管肺曲霉菌病(abpa)引起的恶化的方法。在其他方面,本发明是一种用于缓解呼吸系统疾病和/或慢性肺病(如囊性纤维化、哮喘,尤其是严重的哮喘和严重免疫功能低下的患者)的症状的方法。在另一方面,本发明是一种用于缓解这些患者群体中变应性支气管肺曲霉菌病(abpa)症状的方法。在又另一方面,本发明是一种用于减少炎症,不使用类固醇或减少对甾体治疗的需要的方法。在其他方面,本发明是一种用于改善患有呼吸系统疾病和/或慢性肺病的患者(如囊性纤维化、哮喘,尤其是严重的哮喘和严重免疫功能低下的患者)的肺功能的方法。在另一方面,本发明是一种用于改善患有变应性支气管肺曲霉菌病(abpa)的患者的肺功能的方法。干燥粉末和/或可吸入干燥颗粒可以使用任何适合的方法(如滴注技术)和/或吸入装置(如干燥粉末吸入器(dpi)或定量吸入器(mdi))给予有需要的受试者的呼吸道。许多dpi是可用的,如披露于美国专利号4,995,385和4,069,819中的吸入器、(英国拉夫堡的fisons公司(fisons,loughborough,u.k.))、和(北卡罗来纳州三角科技园的葛兰素史克公司(glaxosmithkline,researchtriangletechnologypark,northcarolina))、(葡萄牙洛里什的好利安公司(hovione,loures,portugal))、(德国的勃林格殷格翰公司(boehringer-ingelheim,germany))、(瑞士的诺华公司(novartis,switzerland)),高阻力、超高阻力和低阻力rs01(意大利的plastiape公司(plastiape,italy))以及本领域技术人员已知的其他dpi。以下科学期刊文章通过引用并入其对以下干燥粉末吸入器(dpi)配置的全面概述:1)单剂量胶囊dpi,2)多剂量泡罩dpi,和3)多剂量贮存器dpi。n.islam,e.gladki,“drypowderinhalers(dpis)—areviewofdevicereliabilityandinnovation[干燥粉末吸入器(dpi)-装置可靠性和创新的综述]”,internationaljournalofpharmaceuticals[国际药学杂志],360(2008):1-11。h.chystyn,“diskusreview[diskus综述]”,internationaljournalofclinicalpractice[国际临床实践杂志],2007年6月,61,6,1022–1036。h.steckel,b.muller,“invitroevaluationofdrypowderinhalersi:drugdepositionofcommonlyuseddevices[干燥粉末吸入器的体外评估i:常用装置的药物沉积]”,internationaljournalofpharmaceuticals[国际药学杂志],154(1997):19-29。一些代表性的基于胶囊的dpi单元是rs-01(意大利的plastiape公司)、(意大利的ph&t公司)、(瑞士的诺华公司)、aerolizer(瑞士的诺华公司)、(瑞士的诺华公司)、(德国的勃林格殷格翰公司)、(马萨诸塞州的civitas公司(civitas,massachusetts))、(缅因州的doseone公司(doseone,maine))、和(罗纳普朗克乐安公司(rhonepoulencrorer))。一些代表性的单位剂量dpi是(明尼苏达州的3m公司(3m,minnesota))、(加利福尼亚州的mannkind公司(mannkind,california))、(加利福尼亚州的mannkind公司)、(英国剑桥的teamconsulting公司(teamconsulting,cambridge,uk))、(山德士公司(sandoz))、(加拿大的trimel生物制药公司(trimelbiopharma,canada))、(葡萄牙洛里什的好利安公司)。一些代表性的基于泡罩的dpi单元是(英国的葛兰素史克公司(gsk))、(gsk)、taper(明尼苏达州的3m公司)、(gsk)、(荷兰的格罗宁根大学(universityofgroningen,netherlands))、(英国的vectura公司(vectura,uk))、(美国明尼苏达州的respirics公司(respirics,minnisota,usa))、(瑞士的诺华公司)、(英国的vectura公司)、(英国的vectura公司)、(美国的microdosetherapeutix公司(microdosetherapeutix,usa))、(印度的cipla公司(cipla,india))、(阿普塔公司(aptar))、(英国的vectura公司)、和(宾夕法尼亚州的迈兰公司(mylan,pennsylvania))。一些代表性的基于贮存器的dpi单元是(vectura公司)、next(chiesi公司)、(orion公司)、(meda公司)、(赛诺菲-安万特公司(sanofi-aventis))、(chiesi公司)、(skyepharma公司)、(vectura公司)、(akela公司)、(瑞典的阿斯利康公司(astrazeneca,sweden))、(瑞典的阿斯利康公司)、和(默克公司(merck)),以及本领域技术人员已知的其他dpi单元。一般来说,吸入装置(例如,dpi)能够在单次吸入中递送最大量的干燥粉末或干燥颗粒,这与泡罩、胶囊(例如,000、00、0e、0、1、2、3和4号,其各自的体积容量为1.37ml、950μl、770μl、680μl、480μl、360μl、270μl和200μl)或吸入器内的含有干燥粉末和/或可吸入干燥颗粒的其他装置的容量有关。优选地,泡罩具有约360微升或更小、约270微升或更小,或更优选地约200微升或更小、约150微升或更小、或约100微升或更小的体积。优选地,胶囊是2号胶囊或4号胶囊。更优选地,胶囊是3号胶囊。因此,递送希望剂量或有效量可能需要两次或更多次吸入。优选地,给予有需要的受试者的每个剂量均含有有效量的可吸入干燥颗粒或干燥粉末,并且使用不多于约4次吸入来给予。例如,每个剂量的干燥粉末或可吸入干燥颗粒可以以单次吸入或2、3、或4次吸入给予。干燥粉末和/或可吸入干燥颗粒优选地使用被动式dpi在单个呼吸激活步骤中给予。当使用这种类型的装置时,受试者吸入的能量使可吸入干燥颗粒分散并将它们吸入呼吸道。适用于本发明方法的干燥粉末和/或可吸入干燥颗粒可以行进通过上气道(即口咽和喉)、下气道,其包括气管,随后分叉成支气管和细支气管,并且通过末端细支气管,其进而分成呼吸性细支气管,然后进入最终呼吸区,肺泡或深肺。在本发明的一个实施例中,可吸入干燥颗粒的大部分质量沉积在深肺中。在本发明的另一个实施例中,递送主要至中央气道。在另一个实施例中,递送至上气道。在优选的实施例中,可吸入干燥颗粒的大部分质量沉积在引导气道中。如果希望或指出,则本文所述的干燥粉末和可吸入干燥颗粒可以与一种或多种其他治疗剂一起给予。其他治疗剂可以通过任何适合的途径给予,如口服、肠胃外(例如,静脉内、动脉内、肌肉内、或皮下注射)、局部、通过吸入(例如,支气管内、鼻内或口腔吸入、鼻内滴液)、直肠、阴道等。可吸入干燥颗粒和干燥粉末可以在给予其他治疗剂之前、基本上同时、或之后给予。优选地,给予干燥粉末和/或可吸入干燥颗粒和其他治疗剂以提供其药理活性的实质上重叠。本文所述的干燥粉末和可吸入干燥颗粒旨在如此吸入,并且本发明排除干燥粉末配制品在制备临时分散体方面的用途。临时分散体是本领域技术人员已知的恰好在使用之前(意味着就在药物给予患者之前)完成的制剂。如本文所用,术语“临时分散体”是指其中溶液或悬浮液不是由制药工业直接生产并以即用形式商业化,而是在制备干燥固体组合物之后的某一时刻(通常在接近给予患者的时刻)制备的所有情况。例证以下实例中使用的材料及其来源在以下列出。氯化钠、硫酸钠、聚山梨醇酯80、油酸、氢氧化铵、甘露醇、乳酸镁、和l-亮氨酸得自西格玛奥德里奇公司(sigma-aldrichco.)(密苏里州圣路易斯)、spectrumchemicals公司(加利福尼亚州加迪纳)、艾普力公司(applichem)(密苏里州马里兰高地)、阿法埃莎公司(alfaaesar)(马萨诸塞州图克斯伯里)、赛默飞世尔公司(thermofisher)(马萨诸塞州沃尔瑟姆)、禾大化学品公司(crodachemicals)(英国东约克郡)或默克公司(德国达姆施塔特)。伊曲康唑得自neuland公司(新泽西州普林斯顿)或sms制药公司(印度telengana州)。两性霉素b得自synbiotics公司(印度艾哈迈达巴德)。ultrapure(ii型astm)水来自水净化系统(马萨诸塞州比勒利卡的密理博公司(milliporecorp.,billerica,ma))、或等同物。方法:悬浮液的几何或体积直径。使用激光衍射技术测定活性剂悬浮液的体积中值直径(x50或dv50),其还可以称为体积中值几何直径(vmgd)。该设备由horibala-950仪器构成,该仪器配备有用于处理和移除样品的自动再循环系统或固定容积的样品比色皿。分散介质的样品,由去离子水或去离子水与小于0.5%的表面活性剂(如聚山梨醇酯80或十二烷基硫酸钠)组成。可以施加超声能量以帮助悬浮液的分散。当激光传输在正确的范围内时,将样品在5的设定下超声处理60秒。然后测量样品并报告颗粒大小分布。干燥粉末的几何或体积直径。使用激光衍射技术测定干燥粉末配制品的体积中值直径(x50或dv50),其还可以称为体积中值几何直径(vmgd)。该设备由helos衍射仪和rodos干燥粉末分散器(新泽西州普林斯顿的sympatec公司(sympatec,inc.,princeton,nj))构成。rodos分散器对颗粒样品施加剪切力,由进入的压缩干燥空气的调节器压力(典型地设定为1.0巴,具有最大孔环压力)控制。可以改变压力设定以改变用于分散粉末的能量的量。例如,可以通过将调节器压力从0.2巴改变到4.0巴来调整分散能量。将粉末样品从微刮刀分配到rodos漏斗中。分散的颗粒行进通过激光束,其中产生的所得衍射光图案典型地使用r1透镜通过一系列检测器收集。然后使用fraunhofer衍射模型将整体衍射图案转换成基于体积的颗粒大小分布,其基础是较小的颗粒以较大的角度衍射光。使用该方法,还根据方程(dv[90]-dv[10)/dv[50]确定分布的跨度。跨度值给出了颗粒大小分布的多分散性的相对指示。通过andersen级联冲击器的空气动力学性能用mk-ii1acfmandersen级联冲击器(英国诺丁汉的科普利科技有限公司(copleyscientificlimited,nottingham,uk))(aci)评估从吸入器装置分散的粉末的空气动力学特性。aci仪器在18℃至25℃的受控环境条件和25%与35%之间的相对湿度(rh)下运行。该仪器由八个阶段构成,这些阶段基于惯性撞击分离气溶胶颗粒。在每个阶段,气溶胶流通过一组喷嘴并撞击在相应的撞击板上。具有足够小惯性的颗粒将继续与气溶胶流进入下一阶段,而剩余的颗粒将撞击该板。在每个连续阶段,气溶胶以更高的速度通过喷嘴,并且空气动力学较小的颗粒在板上被收集。在气溶胶通过最后阶段后,过滤器收集剩余的最小颗粒,称为“最终收集过滤器”。然后可以进行重量分析和/或化学分析以确定颗粒大小分布。短堆叠级联击冲击器,也称为坍塌级联冲击器,也用于使劳动时间减少以评估两个空气动力学颗粒大小切割点。使用该坍塌级联冲击器,除了建立细和粗颗粒分数所需的那些阶段之外的阶段被消除。所使用的撞击技术允许收集两个或八个单独的粉末部分。将胶囊(hpmc,3号;新泽西州皮帕克的capsugelvcaps公司(capsugelvcaps,peapack,nj))用粉末填充至特定重量并置于手持式、呼吸激活的干燥粉末吸入器(dpi)装置,高阻力rs01dpi或超高阻力uhr2dpi(二者均由意大利osnago的plastiape公司(plastiape,osnago,italy)提供)中。将胶囊刺破并将粉末通过以60.0l/min的流速操作的级联冲击器抽出2.0s。在该流速下,八个阶段的校准截止直径是8.6、6.5、4.4、3.3、2.0、1.1、0.5和0.3微米并且对于与短堆叠级联冲击器一起使用的两个阶段,基于andersen级联冲击器,截止直径是5.6微米和3.4微米。通过将过滤器置于该装置中收集这些部分并通过重量测量或hplc上的化学测量确定撞击在它们上的粉末的量。通过下一代冲击器的空气动力学性能。用下一代冲击器(英国诺丁汉的科普利科技有限公司)(ngi)评估从吸入器装置分散的粉末的空气动力学特性。对于利用ngi的测量,ngi仪器在18℃至25℃的受控环境条件和25%与35%之间的相对湿度(rh)下运行。该仪器由七个阶段构成,这些阶段基于惯性撞击分离气溶胶颗粒,并且可以在各种空气流速下操作。在每个阶段,气溶胶流通过一组喷嘴并撞击在相应的撞击表面上。具有足够小惯性的颗粒将继续与气溶胶流进入下一阶段,而剩余的颗粒将撞击该表面。在每个连续阶段,气溶胶以更高的速度通过喷嘴,并且空气动力学较小的颗粒在板上被收集。在气溶胶通过最后阶段后,微孔收集器收集剩余的最小颗粒。然后可以进行重量分析和/或化学分析以确定颗粒大小分布。将胶囊(hpmc,3号;新泽西州皮帕克的capsugelvcaps公司)用粉末填充至特定重量并置于手持式、呼吸激活的干燥粉末吸入器(dpi)装置,高阻力rs01dpi或超高阻力rs01dpi(二者均由意大利osnago的plastiape公司提供)中。将胶囊刺破并将粉末通过以指定的2.0升吸入空气的流速操作的级联冲击器抽出。在指定的流速下,计算这些阶段的截止直径。通过将浸湿的过滤器置于该装置中收集这些部分并通过hplc上的化学测量确定撞击在它们上的粉末的量。细颗粒剂量。细颗粒剂量表示特定大小范围内的一种或多种治疗剂的质量,并且可以用于预测将到达呼吸道中某个区域的质量。细颗粒剂量可以通过aci或ngi通过重量分析或化学地测量。如果通过重量分析测量,由于假定干燥颗粒是均匀的,因此每个阶段和收集过滤器上的粉末质量可以乘以配制品中治疗剂的分数以确定治疗剂的质量。如果化学地测量,则收集、分离来自每个阶段或过滤器的粉末,并例如在hplc上测定以确定治疗剂的含量。计算以指定流速沉积在每个阶段上的累积质量,并且插入对应于5.0微米直径颗粒的累积质量。包含在一个或多个胶囊中的致动到冲击器中的单剂量粉末的累积质量等于小于5.0微米的细颗粒剂量(fpd<5.0微米)。质量中值空气动力学直径。使用通过andersen级联冲击器(aci)获得的信息确定质量中值空气动力学直径(mmad)。计算每个阶段的阶段截止直径下的累积质量,并通过回收的粉末剂量归一化。然后通过包含第50百分位数的阶段截止直径的线性插值来计算粉末的mmad。测量mmad的替代方法是使用下一代冲击器(ngi)。与aci一样,用以下计算mmad:计算每个阶段的阶段截止直径下的累积质量,并通过回收的粉末剂量归一化。然后通过包含第50百分位数的阶段截止直径的线性插值来计算粉末的mmad。发射的几何或体积直径。使用激光衍射技术通过spraytec衍射仪(马尔文公司(malvern,inc.))测定粉末从干燥粉末吸入器发射后的体积中值直径(dv50),其还可以称为体积中值几何直径(vmgd)。将粉末填充到3号胶囊(v-caps,capsugel公司)中,并置于基于胶囊的干燥粉末吸入器(rs01型7高阻力,意大利的plastiape公司)或dpi中,并将dpi密封在圆筒内。将圆筒连接到正压空气源,同时用质量流量计测量通过系统的稳定气流,并且用定时器控制的电磁阀控制其持续时间。使干燥粉末吸入器的出口暴露于室压,并且所得气溶胶射流在其通过真空提取器捕获之前通过其开放式工作台配置中衍射颗粒分析仪(spraytec)的激光器。使用电磁阀启动通过系统的稳定空气流速。通过dpi典型地以60l/min抽出稳定的空气流速持续设定的持续时间,典型地为2秒。可替代地,通过dpi抽出的空气流速有时以15l/min、20l/min、或30l/min运行。由软件基于光电探测器上测量的散射图案计算所得气溶胶的几何颗粒大小分布,其中样品典型地在1000hz下在吸入过程中取得。然后平均吸入过程中所测量的dv50、gsd、fpf<5.0μm。发射剂量(ed)是指在烧制或分散事件后离开适合的吸入器装置的治疗剂的质量。使用基于unitedstatespharmacopeiaconvention[美国药典公约],罗克维尔市,马里兰州,第13版,222-225,2007的usp第601节气溶胶,计量剂量吸入器和干燥粉末吸入器,递送剂量均匀性,从干燥粉末吸入器取样递送剂量的方法来确定ed。使用rs01hr吸入器以4kpa的压降和60lpm的典型流速或使用uhr2rs01以4kpa的压降和39lpm的典型流速分散胶囊的内容物。将发射的粉末收集在过滤器支架取样装置中的过滤器上。将取样装置用适合的溶剂(如水)冲洗,并使用hplc方法分析。对于重量分析,使用较短长度的过滤器支架取样装置来减少装置中的沉积,并且在之前和之后称重过滤器以确定从dpi递送到过滤器的粉末质量。然后基于递送粉末中的治疗剂含量计算治疗剂的发射剂量。发射剂量可以报告为从dpi递送的治疗剂的质量或填充剂量的百分比。热重分析:使用q500模型或discovery模型热重分析仪(特拉华州纽卡斯尔的德州仪器公司(tainstruments,newcastle,de))进行热重分析(tga)。将样品置于开口铝dsc盘或随后在测试之前自动打开的密封铝dsc盘中。皮重先前由仪器记录。采用以下方法:从环境温度(约35℃)至200℃,速率为5.00℃/min。据报道,重量损失为高达140℃的温度的函数。tga允许计算干燥粉末中挥发性化合物的含量。当单独使用水或将水与挥发性溶剂结合使用时,通过tga的重量损失是对水含量的良好估计。x射线粉末衍射:通过粉末x射线衍射(pxrd)评估配制品的结晶特征。在粉末x射线衍射仪(具有linxeye检测器的d8discover;马萨诸塞州比勒利卡的布鲁克公司(brukercorporation,billerica,ma)或等同物)中使用具有1.5418a的cux射线管以数据累积时间1.2秒/步和0.02°2θ的步长在5至45°2θ的扫描范围内分析20-30mg材料样品。使用hplc测量的伊曲康唑含量/纯度。已开发了利用与紫外(uv)检测器偶联的反相c18柱的高效液相色谱(hplc)方法,用于pur1900配制品的鉴定、本体含量、测定、cupmd和杂质分析。将反相柱平衡至30℃,并且将自动进样器设定至5℃。将流动相(在ph2.0下为20mm磷酸二氢钠(流动相a)和乙腈(流动相b))用于在19.5分钟的运行时间内梯度洗脱从比率59:41(a:b)至5:95(a:b)。通过uv在258nm处进行检测,并且注射体积是10μl。相对于标准曲线定量粉末中的伊曲康唑含量。通过将pur1900样品中杂质峰的保留时间与掺杂有杂质a的伊曲康唑usp杂质混合参考标准物的保留时间进行比较来确定已知杂质a、b、c、d、e、f和g的鉴定(在欧洲药典专论01/2011:1335中示出)。通过相对于伊曲康唑主峰的保留时间和高于检测限(lod)的面积来鉴定和量化未知杂质。相对于伊曲康唑峰,通过面积百分比测量所有杂质。颗粒大小减小。可以使用本领域技术人员熟悉的许多技术来调整结晶活性剂的颗粒大小分布,包括但不限于高压均化、高剪切均化、喷射研磨、针磨、微流化、或湿磨(也称为球磨、珍珠磨或珠磨)。湿磨经常是优选的,因为它能够实现宽范围的颗粒大小分布,包括纳米(<1μm)大小域中的那些。使用低能量湿磨的颗粒大小减小。用于减小活性剂颗粒大小的一种技术是通过低能量湿磨(也称为辊磨或罐磨)。在抗溶剂中制备活性剂的悬浮液,该抗溶剂可以是水,或其中活性剂不明显可溶的任何溶剂。然后将稳定剂(可以是但不限于非离子表面活性剂或两亲聚合物)与研磨介质一起加入悬浮液中,该研磨介质可以是但不限于具有高耐磨性且直径大小在从0.03至0.70毫米范围内的球形。然后使用罐磨机(美国俄亥俄州东巴勒斯坦城的usstoneware公司(usstoneware,eastpalestine,ohusa))旋转含有悬浮液的容器,同时定期取样以评估颗粒大小(la-950,日本京都的horiba公司(horiba,kyoto,japan))。当颗粒大小充分减小时,或当达到最小颗粒大小时,将悬浮液通过筛子过滤以除去研磨介质,并回收产物。使用高能量湿磨的颗粒大小减小。用于减小活性剂颗粒大小的另一种技术是通过使用转子-定子或搅拌介质研磨机的高能量湿磨。在抗溶剂中制备活性剂的悬浮液,该抗溶剂可以是水,或其中活性剂不明显可溶的任何溶剂。然后将稳定剂(可以是但不限于非离子表面活性剂或两亲聚合物)与研磨介质一起加入悬浮液中,该研磨介质可以是但不限于具有高耐磨性且直径大小在从0.03至0.70毫米范围内的球形。然后将悬浮液加入研磨机中,该研磨机可以以分批或再循环模式操作。该方法包括在研磨室内搅拌悬浮液和研磨介质,这增加了输入系统的能量并加速了颗粒大小的减小过程。研磨室和再循环容器有夹套并主动冷却以避免产物中的温度升高。在该过程中控制悬浮液的搅拌速率和再循环速率。定期取样以评估颗粒大小(la-950,日本京都的horiba公司)。当颗粒大小充分减小时,或当达到最小颗粒大小时,将悬浮液从研磨机排出。使用微流化的颗粒大小减小。用于减小活性剂的颗粒大小分布的另一种技术是通过微流化。基于微流化器的处理是用于液体和固体的颗粒大小减小的高剪切湿处理单元操作。该单元可以配置有各种相互作用室,这些室是具有特定孔和通道设计的圆柱形模块,流体在高压下通过这些孔和通道以控制剪切速率。产物通过入口贮存器进入该单元,并通过高压泵以高达400m/sec的速度压入固定几何形状的相互作用室。然后,如果需要,将其有效冷却并收集在输出容器中。可以根据需要重复该过程(例如多次“通过”)以实现颗粒大小目标。通过激光衍射(la-950,日本京都的horiba公司)定期监测活性剂的颗粒大小。当颗粒大小充分减小时,或当达到最小颗粒大小时,从该单元回收悬浮液。使用喷射研磨的颗粒大小减小。用于减小活性剂的颗粒大小分布的另一种技术是通过喷射研磨。喷射研磨机利用流体能量(压缩空气或气体)在没有活动部件的单个室中进行研磨和分类。通过高压空气活化,颗粒在浅的研磨室中被加速成高速旋转。当颗粒彼此影响时,它们的大小被减小。离心力在研磨旋转区域中保持较大的颗粒,直至它们实现所希望的细颗粒大小。向心力将所希望的颗粒拖向静态分级器,在实现正确的颗粒大小时允许它们离开。通过改变进料速率和推进剂压力来控制最终颗粒大小。用于喷雾干燥的液体原料制备。喷雾干燥均匀颗粒需要将感兴趣的成分溶解在溶液中或悬浮在均匀且稳定的悬浮液中。原料可以利用水,或水和其他可混溶溶剂(如醇或酮)的组合,在溶液的情况下作为溶剂,或在悬浮液的情况下作为连续相。各种配制品的原料通过将可溶性组分溶解在一种或多种所希望溶剂中,随后在混合时将表面活性剂稳定的含有活性剂的悬浮液分散在所得溶液中来制备,尽管该方法不限于该特定操作顺序。使用niro喷雾干燥器的喷雾干燥。利用niromobileminor喷雾干燥器(马里兰州哥伦比亚的gea工程工艺公司(geaprocessengineeringinc.,columbia,md))通过喷雾干燥产生干燥粉末,其中粉末收集来自旋风分离器、产物过滤器或二者。使用来自niro(马里兰州哥伦比亚的gea工程工艺公司)的并流双流体喷嘴或具有气盖67147和流体盖2850ss的sprayingsystems(伊利诺伊州的carolstream公司)1/4j双流体喷嘴进行液体进料的雾化,尽管其他双流体喷嘴设置也是可能的。在一些实施例中,双流体喷嘴可以是内部混合设置或外部混合设置。另外的雾化技术包括旋转雾化或压力喷嘴。使用齿轮泵(伊利诺伊州弗农希尔斯的cole-parmer仪器公司(cole-parmerinstrumentcompany,vernonhills,il))将液体进料直接进料到双流体喷嘴中或静态混合器(纽约哈帕克的查尔斯罗斯父子公司(charlesross&soncompany,hauppauge,ny))中紧接着引入双流体喷嘴中。另外的液体进料技术包括从加压容器进料。氮气或空气可以用作干燥气体,条件是空气中的水分在使用前至少部分地除去。加压氮气或空气可以用作双流体喷嘴的雾化气体进料。干燥气体入口温度可以在从70℃至300℃范围内,并且出口温度从30℃至120℃,其中液体原料速率为10ml/min至100ml/min。供应双流体雾化器的气体可以根据喷嘴选择而变化,并且对于niro并流双流体喷嘴可以在从5kg/hr至50kg/hr范围内,或者对于sprayingsystems1/4j双流体喷嘴可以在从30g/min至150g/min范围内。可以设定雾化气体速率以实现一定的气体与液体质量比,这直接影响所产生的液滴大小。干燥转筒内的压力可以在从+3“wc至-6“wc范围内。喷雾干燥的粉末可以收集在旋风分离器出口处的容器中、收集在筒式或袋式过滤器上,或者收集在旋风分离器和筒式或袋式过滤器二者中。使用büchi喷雾干燥器的喷雾干燥。通过在büchib-290minispraydryer(瑞士弗拉维尔的步琪实验室技术服务有限公司)上喷雾干燥制备干燥粉末,其中粉末收集来自标准或高性能旋风分离器。该系统以空气或氮气作为干燥和雾化气体以开环(单程)模式运行。当使用空气运行时,该系统使用büchib-296除湿器以确保用于喷雾干燥的空气的稳定温度和湿度。此外,当房间内的相对湿度超过30%rh时,外部lg除湿器(型号49007903,新泽西州恩格尔伍德克利夫斯的lg电子公司(lgelectronics,englewoodcliffs,nj))不断地运行。当使用氮气运行时,使用加压的氮源。此外,调节系统的吸气器以将系统压力维持在-2.0”水柱。液体进料的雾化利用具有1.5mm直径的büchi双流体喷嘴或具有0.5mm液体插入物的schlick970-0雾化器(德国科堡的düsen-schlick有限公司(düsen-schlickgmbh,coburg,germany))。工艺气体的入口温度可以在从100℃至220℃范围内,并且出口温度从30℃至120℃,其中液体原料流速为3ml/min至10ml/min。对于büchi双流体喷嘴,双流体雾化气体在从25mm至45mm(300lph至530lph)范围内,并且对于schlick雾化器,雾化气体压力在0.3巴以上。吸气器比率在从50%至100%范围内。稳定性评估:在2℃-8℃、25℃/60%rh并且当材料数量允许时,如国际协调会议(ich)q1指导中详述在40℃/75%rh下评估所选择配制品的物理化学稳定性和气溶胶性能。将稳定性样品储存在校准室(达尔文钱伯斯公司(darwinchamberscompany)型号ph024和ph074,密苏里州圣路易斯)中。将本体粉末样品称入琥珀色玻璃小瓶中,在30%rh下密封,并用二氧化硅干燥剂(2.0g,纽约布法罗的multisorbtechnologies公司(multisorbtechnologies,buffalo,ny))感应密封在铝袋(drishield3000,明尼苏达州的3m公司)中。另外,为了评估配制品在胶囊中的稳定性,将目标质量的粉末用手称入3号hpmc胶囊(新泽西州皮帕克的capsugelvcaps公司)中,该胶囊在30%rh下具有+/-0.2mg耐受性。然后将填充的胶囊等分到高密度聚乙烯(hdpe)瓶中,并用二氧化硅干燥剂感应密封在铝袋中。实例1.含有硫酸钠/甘露醇的聚山梨醇酯80稳定的纳米结晶伊曲康唑的干燥粉末配制品a.粉末制备。通过将11.662g伊曲康唑(neuland公司批号iti0114005)在103.789g水和1.1662g聚山梨醇酯80(spectrum批号2di0112)中配混来制备纳米结晶伊曲康唑。然后将129.625g500μm聚苯乙烯研磨介质(密歇根州米德兰的陶氏化学公司(dowchemical,midlandmi))加入悬浮液中,并且将悬浮液以1000rpm研磨一小时接着以1500rpm研磨30分钟,然后收集。研磨悬浮液的最终中值颗粒大小(dv(50))是124nm。制备原料溶液并将其用于制造由纳米结晶伊曲康唑、聚山梨醇酯80和其他另外的赋形剂组成的干燥粉末。目标是在干基上20wt%和50wt%伊曲康唑的药物负载。用于喷雾干燥颗粒的原料溶液如下制备。将所需量的水称入适当大小的玻璃容器中。将赋形剂加入水中,并且搅拌溶液直至视觉上澄清。然后将含有伊曲康唑的悬浮液加入赋形剂溶液中并搅拌直至视觉上均匀。然后喷雾干燥原料。在喷雾干燥的同时搅拌原料。原料质量是83.3g,这支持15分钟的制造活动。表2列出了用于制备干燥粉末的原料组分。表2:原料组成和干燥粉末组成(w/w),干基用旋风分离器粉末收集,通过在büchib-290minispraydryer(瑞士弗拉维尔的步琪实验室技术服务有限公司)上喷雾干燥由这些原料制造配制品i和ii的干燥粉末。该系统使用氮气作为干燥和雾化气体以开环(单程)模式运行。液体进料的雾化利用带有1.5mm盖子和0.7液体尖端的büchi喷嘴。调节系统的吸气器以将系统压力维持在-2.0”水柱。遵循以下喷雾干燥条件以制造干燥粉末。对于配制品i和ii,液体原料固体浓度是3.0wt%,工艺气体入口温度是117℃至119℃,工艺气体出口温度是50℃,干燥气体流速是17.0kg/hr,雾化气体流速是30.4g/min,雾化气体和液体原料流速是6.0ml/min。所得干燥粉末配制品报告于下表3中。表3b.粉末表征。两种配制品的本体颗粒大小表征见于表4。配制品i和ii的1巴下跨度分别是1.83和1.67,表明相对较窄的大小分布。配制品i和ii的1巴/4巴分散性比分别是1.07和1.12,表明它们相对独立于分散能量,这是一种希望的特征,其允许在一系列分散能量内产生类似的颗粒分散。表4:本体颗粒大小测量两种配制品以60升/分钟(lpm)和20lpm模拟的患者流速测量和/或计算的几何颗粒大小和胶囊发射的粉末质量(cepm)并报告在表5中。从60lpm至20lpm的cepm和几何大小的微小变化表明干燥粉末配制品相对独立于患者吸入流速,这表明以不同流速呼吸的患者将接受相对类似的治疗剂量。表5:发射颗粒大小用八阶段anderson级联冲击器(aci-8)测量和/或计算的空气动力学颗粒大小、细颗粒分数和细颗粒剂量报告于表6中。配制品i和ii二者的细颗粒剂量均表明高百分比的填充到胶囊中的标称剂量到达冲击器阶段(分别是38.8%和37.1%),并且因此预测将递送到肺部。配制品i和ii的mmad分别是3.59微米和3.17微米,表明在中央和引导气道中沉积。表6:空气动力学颗粒大小通过tga测量配制品i和ii的重量损失,并且发现分别为0.48%和0.15%。用hplc-uv测量配制品i和ii的伊曲康唑含量,并且分别为102.9%和103.1%。通过xrd评估配制品i和ii的结晶度。在两种配制品中均观察到伊曲康唑的衍射图案,表明研磨或喷雾干燥过程不影响伊曲康唑的固态。在图案中观察到的另外的峰对应于配制品中的另外的赋形剂。(图1)在2℃-8℃和25℃/60%rh下储存6个月后,确定配制品i和ii是稳定的。实例2.含有氯化钠/亮氨酸的聚山梨醇酯80稳定的纳米结晶伊曲康唑的干燥粉末配制品a.粉末制备。通过将11.662g伊曲康唑(neuland公司批号iti0114005)在103.789g水和1.1662g聚山梨醇酯80(spectrum批号2di0112)中配混来制备纳米结晶伊曲康唑。然后将129.625g500μm聚苯乙烯研磨介质(密歇根州米德兰的陶氏化学公司)加入悬浮液中,并且将悬浮液以1000rpm研磨一小时接着以1500rpm研磨30分钟,然后收集。研磨悬浮液的最终中值颗粒大小(dv(50))是124nm。制备原料溶液并将其用于制造由纳米结晶伊曲康唑、聚山梨醇酯80和其他另外的赋形剂组成的干燥粉末。目标是在干基上20wt%和50wt%伊曲康唑的药物负载。用于喷雾干燥颗粒的原料溶液如下制备。将所需量的水称入适当大小的玻璃容器中。将赋形剂加入水中,并且搅拌溶液直至视觉上澄清。然后将含有伊曲康唑的悬浮液加入赋形剂溶液中并搅拌直至视觉上均匀。然后喷雾干燥原料。在喷雾干燥的同时搅拌原料。原料质量是83.3g,这支持15分钟的制造活动。表7列出了用于制备干燥粉末的原料组分。表7:原料组成用旋风分离器粉末收集,通过在büchib-290minispraydryer(瑞士弗拉维尔的步琪实验室技术服务有限公司)上喷雾干燥由这些原料制造配制品i和ii的干燥粉末。该系统使用氮气作为干燥和雾化气体以开环(单程)模式运行。液体进料的雾化利用带有1.5mm盖子和0.7液体尖端的büchi喷嘴。调节系统的吸气器以将系统压力维持在-2.0”水柱。遵循以下喷雾干燥条件以制造干燥粉末。对于配制品i和ii,液体原料固体浓度是3.0%,工艺气体入口温度是138℃至141℃,工艺气体出口温度是60℃,干燥气体流速是17.0kg/hr,雾化气体流速是30.4g/min,雾化气体和液体原料流速是6.0ml/min。所得干燥粉末配制品报告于表8中。表8:干燥粉末组成,干基配制品干燥粉末组成(w/w),干基iii20%伊曲康唑、62.4%氯化钠、15.6%亮氨酸、2%聚山梨醇酯80iv50%伊曲康唑、36%氯化钠、9%亮氨酸、5%聚山梨醇酯80b.粉末表征。两种配制品的本体颗粒大小表征见于表9。配制品iii和iv的1巴下跨度分别是1.76和1.86,表明相对较窄的大小分布。配制品iii和iv的1巴/4巴分散性比分别是1.19和1.05,表明它们相对独立于分散能量,这是一种希望的特征,其允许在一系列分散能量内产生类似的颗粒分散。表9:本体颗粒大小测量两种配制品以60升/分钟(lpm)和20lpm模拟的患者流速测量和/或计算的几何颗粒大小和胶囊发射的粉末质量(cepm)并报告在表10中。从60lpm至20lpm的cepm和几何大小的微小变化表明干燥粉末配制品相对独立于患者吸入流速,这表明以不同流速呼吸的患者将接受相对类似的治疗剂量。表10:发射颗粒大小用八阶段anderson级联冲击器(aci-8)测量和/或计算的空气动力学颗粒大小、细颗粒分数和细颗粒剂量报告于表11中。配制品iii和iv二者的细颗粒剂量均表明高百分比的填充到胶囊中的标称剂量到达冲击器阶段(分别是55.5%和49.4%),并且因此预测将递送到肺部。配制品iii和iv的mmad分别是3.14微米和3.30微米,表明在中央和引导气道中沉积。表11:空气动力学颗粒大小通过tga测量配制品iii和iv的重量损失,并且发现分别为0.15%和0.08%。用hplc-uv测量配制品iii和iv的伊曲康唑含量,并且分别为103.7%和104.9%。通过xrd评估配制品iii和iv的结晶度。在两种配制品中均观察到伊曲康唑的衍射图案,表明研磨或喷雾干燥过程不影响伊曲康唑的固态。在图案中观察到的另外的峰对应于配制品中的另外的赋形剂。(图2)在2℃-8℃和25℃/60%rh下储存6个月后,确定配制品iii和iv是稳定的。实例3.含有乳酸镁/亮氨酸的聚山梨醇酯80稳定的纳米结晶伊曲康唑的干燥粉末配制品a.粉末制备。通过将11.662g伊曲康唑(neuland公司批号iti0114005)在103.789g水和1.1662g聚山梨醇酯80(spectrum批号2di0112)中配混来制备纳米结晶伊曲康唑。然后将129.625g500μm聚苯乙烯研磨介质(密歇根州米德兰的陶氏化学公司)加入悬浮液中,并且将悬浮液以1000rpm研磨一小时接着以1500rpm研磨30分钟,然后收集。研磨悬浮液的最终中值颗粒大小(dv(50))是124nm。制备原料溶液并将其用于制造由纳米结晶伊曲康唑、聚山梨醇酯80和其他另外的赋形剂组成的干燥粉末。目标是在干基上20wt%和50wt%伊曲康唑的药物负载。用于喷雾干燥颗粒的原料溶液如下制备。将所需量的水称入适当大小的玻璃容器中。将赋形剂加入水中,并且搅拌溶液直至视觉上澄清。然后将含有伊曲康唑的悬浮液加入赋形剂溶液中并搅拌直至视觉上均匀。然后喷雾干燥原料。在喷雾干燥的同时搅拌原料。原料质量是83.3g,这支持15分钟的制造活动。表12列出了用于制备干燥粉末的原料组分。表12:原料组成用旋风分离器粉末收集,通过在büchib-290minispraydryer(瑞士弗拉维尔的步琪实验室技术服务有限公司)上喷雾干燥由这些原料制造配制品v和vi的干燥粉末。该系统使用氮气作为干燥和雾化气体以开环(单程)模式运行。液体进料的雾化利用带有1.5mm盖子和0.7液体尖端的büchi喷嘴。调节系统的吸气器以将系统压力维持在-2.0”水柱。遵循以下喷雾干燥条件以制造干燥粉末。对于配制品v和vi,液体原料固体浓度是3.0%,工艺气体入口温度是171℃至173℃,工艺气体出口温度是80℃,干燥气体流速是17.0kg/hr,雾化气体流速是30.4g/min,雾化气体和液体原料流速是6.0ml/min。所得干燥粉末配制品报告于表13中。表13:干燥粉末组成,干基配制品干燥粉末组成(w/w),干基v20%伊曲康唑、66.3%乳酸镁、11.7%亮氨酸、2%聚山梨醇酯80vi50%伊曲康唑、38.25%乳酸镁、6.75%亮氨酸、5%聚山梨醇酯80b.粉末表征。两种配制品的本体颗粒大小表征见于表14。配制品v和vi的1巴下跨度分别是1.70和1.83,表明相对较窄的大小分布。配制品v和vi的1巴/4巴分散性比分别是1.02和1.05,表明它们相对独立于分散能量,这是一种希望的特征,其允许在一系列分散能量内产生类似的颗粒分散。表14:本体颗粒大小测量两种配制品以60升/分钟(lpm)和20lpm模拟的患者流速测量和/或计算的几何颗粒大小和胶囊发射的粉末质量(cepm)并报告在表15中。从60lpm至20lpm的cepm和几何大小的微小变化表明干燥粉末配制品相对独立于患者吸入流速,这表明以不同流速呼吸的患者将接受相对类似的治疗剂量。表15:发射颗粒大小用八阶段anderson级联冲击器(aci-8)测量和/或计算的空气动力学颗粒大小、细颗粒分数和细颗粒剂量报告于表16中。配制品v和vi二者的细颗粒剂量均表明高百分比的填充到胶囊中的标称剂量到达冲击器阶段(分别是39.6%和44.6%),并且因此预测将递送到肺部。配制品v和vi的mmad分别是3.97微米和3.42微米,表明在中央和引导气道中沉积。表16:空气动力学颗粒大小通过tga测量配制品v和vi的重量损失,并且发现分别为5.157%和3.087%。用hplc-uv测量配制品v和vi的伊曲康唑含量,并且分别为99.7%和100.6%。通过xrd评估配制品v和vi的结晶度。在两种配制品中均观察到伊曲康唑的衍射图案,表明研磨或喷雾干燥过程不影响伊曲康唑的固态。在图案中观察到的另外的峰对应于配制品中的另外的赋形剂。(图3)在2℃-8℃和25℃/60%rh下储存6个月后,确定配制品v和vi是稳定的。实例4.含有硫酸钠/亮氨酸的油酸稳定的纳米结晶伊曲康唑的干燥粉末配制品a.粉末制备。通过将11.646g伊曲康唑(neuland公司批号iti0114005)在104.233g水、0.582g油酸(禾大公司000705097)、和9.44g10%氢氧化铵中配混来制备纳米结晶伊曲康唑。然后将129.625g500μm聚苯乙烯研磨介质(密歇根州米德兰的陶氏化学公司)加入悬浮液中,并且将悬浮液以1000rpm研磨一小时并且接着以1500rpm再研磨一小时,然后收集。研磨悬浮液的最终中值颗粒大小(dv(50))是120nm。制备原料溶液并将其用于制造由纳米结晶伊曲康唑、油酸和其他另外的赋形剂组成的干燥粉末。目标是在干基上50wt%和70wt%伊曲康唑的药物负载。用于喷雾干燥颗粒的原料溶液如下制备。将所需量的水称入适当大小的玻璃容器中。将赋形剂加入水中,并且搅拌溶液直至视觉上澄清。然后将含有伊曲康唑的悬浮液加入赋形剂溶液中并搅拌直至视觉上均匀。然后喷雾干燥原料。在喷雾干燥的同时搅拌原料。原料体积范围为从100至193.3g,这支持了从16至34分钟的制造活动。表17列出了用于制备干燥粉末的原料组分。表17:原料组成用旋风分离器粉末收集,通过在büchib-290minispraydryer(瑞士弗拉维尔的步琪实验室技术服务有限公司)上喷雾干燥由这些原料制造配制品vii和viii的干燥粉末。该系统使用氮气作为干燥和雾化气体以开环(单程)模式运行。液体进料的雾化利用带有1.5mm盖子和0.7液体尖端的büchi喷嘴。调节系统的吸气器以将系统压力维持在-2.0”水柱。遵循以下喷雾干燥条件以制造干燥粉末。对于配制品vii和viii,液体原料固体浓度是3.0%,工艺气体入口温度是131℃至133℃,工艺气体出口温度是60℃,干燥气体流速是17.0kg/hr,雾化气体流速是30.4g/min(1.824kg/hr),并且液体原料流速是6.0ml/min。所得干燥粉末配制品报告于表18中。表18:干燥粉末组成,干基配制品干燥粉末组成(w/w),干基vii50%伊曲康唑、33.25%硫酸钠、14.25%亮氨酸、2.5%油酸viii70%伊曲康唑、13.25%硫酸钠、13.25%亮氨酸、3.5%油酸b.粉末表征。两种配制品的本体颗粒大小表征见于表19。配制品vii和viii的1巴下跨度分别是1.94和1.81,表明相对较窄的大小分布。配制品vii和viii的1巴/4巴分散性比分别是1.22和1.11,表明它们相对独立于分散能量,这是一种希望的特征,其允许在一系列分散能量内产生类似的颗粒分散。表19:本体颗粒大小测量两种配制品以60升/分钟(lpm)和20lpm模拟的患者流速测量和/或计算的几何颗粒大小和胶囊发射的粉末质量(cepm)并报告在表20中。从60lpm至20lpm的cepm和几何大小的微小变化表明干燥粉末配制品相对独立于患者吸入流速,这表明以不同流速呼吸的患者将接受相对类似的治疗剂量。表20:发射颗粒大小用八阶段anderson级联冲击器(aci-8)测量和/或计算的空气动力学颗粒大小、细颗粒分数和细颗粒剂量报告于表21中。配制品vii和viii二者的细颗粒剂量均表明高百分比的填充到胶囊中的标称剂量到达冲击器阶段(分别是56.0%和52.6%),并且因此预测将递送到肺部。配制品vii和viii的mmad分别是2.77微米和3.08微米,表明在中央和引导气道中沉积。表21:空气动力学颗粒大小通过tga测量配制品vii和viii的重量损失,并且发现分别为0.47%和0.33%。用hplc-uv测量配制品vii和viii的伊曲康唑含量,并且分别为101.5%和101.4%。通过xrd评估配制品vii和viii的结晶度。在两种配制品中均观察到伊曲康唑的衍射图案,表明研磨或喷雾干燥过程不影响伊曲康唑的固态。在图案中观察到的另外的峰对应于配制品中的另外的赋形剂。(图4)在2℃-8℃和25℃/60%rh下储存6个月后,确定配制品vii和viii是稳定的。实例5.参考液体纳米结晶和微晶伊曲康唑配制品制备了结晶微粒伊曲康唑的液体配制品。配制品ix是伊曲康唑与聚山梨醇酯80的微悬浮液。液体中的伊曲康唑浓度是5mg/ml。伊曲康唑与聚山梨醇酯80的比率是10:1(wgt/wgt)。伊曲康唑晶体的中值大小是1600纳米。配制品x是伊曲康唑与聚山梨醇酯80的纳米悬浮液。液体中的伊曲康唑浓度是5mg/ml。伊曲康唑与聚山梨醇酯80的比率是10:1(wgt/wgt)。伊曲康唑晶体的中值大小是132纳米。实例6.含有硫酸钠/亮氨酸的油酸稳定的纳米结晶伊曲康唑的干燥粉末配制品a.粉末制备。通过将30.374g伊曲康唑(neuland公司iti0714011)在87.018g水、1.519g油酸(croda公司000705097)、和2.585g氢氧化铵(acros公司b0522464)中配混来制备纳米结晶伊曲康唑。然后将129.625g500μm聚苯乙烯研磨介质(密歇根州米德兰的陶氏化学公司)加入悬浮液中,并且将悬浮液以1800rpm研磨两小时,然后收集。研磨悬浮液的最终中值颗粒大小(dv(50))是124nm。制备原料溶液并将其用于制造由纳米结晶伊曲康唑、油酸和其他另外的赋形剂组成的干燥粉末。目标是在干基上50wt%伊曲康唑的药物负载。用于喷雾干燥颗粒的原料溶液如下制备。将所需量的水称入适当大小的玻璃容器中。将赋形剂加入水中,并且搅拌溶液直至视觉上澄清。然后将含有伊曲康唑的悬浮液加入赋形剂溶液中并搅拌直至视觉上均匀。然后将原料喷雾干燥。原料质量是1219.4g,这支持大约3.5小时的制造活动。表22列出了用于制备干燥粉末的原料组分。表22:原料组成用旋风分离器粉末收集,通过在büchib-290minispraydryer(瑞士弗拉维尔的步琪实验室技术服务有限公司)上喷雾干燥由该原料制造配制品xi的干燥粉末。该系统使用氮气作为干燥和雾化气体以开环(单程)模式运行。液体进料的雾化利用带有1.5mm盖子和0.7液体尖端的büchi喷嘴。调节系统的吸气器以将系统压力维持在-2.0”水柱。遵循以下喷雾干燥条件以制造干燥粉末。对于配制品xi,液体原料固体浓度是3.0%,工艺气体入口温度是129℃至132℃,工艺气体出口温度是60℃,干燥气体流速是17.0kg/hr,雾化气体流速是30.4g/min,并且液体原料流速是6.0ml/min。所得干燥粉末配制品报告于表23中。表23:干燥粉末组成,干基配制品干燥粉末组成(w/w),干基xi50%伊曲康唑、35%硫酸钠、12.5%亮氨酸、2.5%油酸b.粉末表征。该配制品的本体颗粒大小表征见于表24。配制品xi的1巴下跨度是2.77,表明相对较窄的大小分布。配制品xi的1巴/4巴分散性比是1.28,表明颗粒大小相对独立于分散能量,这是一种希望的特征,其允许在一系列分散能量内产生类似的分散。表24:本体颗粒大小测量该配制品以60升/分钟(lpm)和20lpm模拟的患者流速测量和/或计算的几何颗粒大小和胶囊发射的粉末质量(cepm)并报告在表25中。从60lpm至20lpm的cepm和几何大小的微小变化表明干燥粉末配制品相对独立于患者吸入流速,这表明以不同流速呼吸的患者将接受相对类似的治疗剂量。表25:发射颗粒大小用下一代冲击器(ngi)测量和/或计算的空气动力学颗粒大小、细颗粒分数和细颗粒剂量报告于表26中。配制品xi的细颗粒剂量表明高百分比的填充到胶囊中的标称剂量到达冲击器阶段(42%),并且因此预测将递送到肺部。配制品xi的mmad是3.37微米,表明在中央和引导气道中沉积。表26:空气动力学颗粒大小通过tga测量配制品xi的重量损失,并且发现为0.31%。用hplc-uv测量配制品xi的伊曲康唑含量并且是99.7%。通过xrd评估配制品xi的结晶度。在该配制品中观察到伊曲康唑的衍射图案,表明研磨或喷雾干燥过程不影响伊曲康唑的固态。在图案中观察到的另外的峰对应于配制品中的另外的赋形剂。(图5)实例7.含有硫酸钠/亮氨酸的具有不同颗粒大小的聚山梨醇酯80稳定的结晶伊曲康唑的干燥粉末配制品a.粉末制备。通过将30.090g伊曲康唑(neuland公司iti0114005和iti0714011)在87.262g水和3.009g聚山梨醇酯80中配混来制备用于配制品xii的纳米结晶伊曲康唑。然后将129.625g500μm聚苯乙烯研磨介质(密歇根州米德兰的陶氏化学公司)加入悬浮液中,并且将悬浮液以1800rpm研磨一小时,然后收集。研磨悬浮液的最终中值颗粒大小(dv(50))是132nm。下文将该过程称为“湿磨过程#1”。将用于配制品xiii的纳米结晶伊曲康唑制备为在去离子水中包含10wt%伊曲康唑和1.0wt%聚山梨醇酯80的悬浮液。通过磁力搅拌棒将聚山梨醇酯80溶解在89.0%di水中,然后还用磁力搅拌棒缓慢加入伊曲康唑。一旦所有伊曲康唑悬浮,则将配制品在m-110pmicrofluidizer处理器上以30,000psi处理120次,使用冰水冷却盘管以在处理过程中冷却材料。研磨悬浮液的最终中值颗粒大小(dv(50))是198nm。下文将该过程称为“微流体过程#1”。通过将30.090g伊曲康唑(neuland公司iti0114005)在87.26195g水和3.009g聚山梨醇酯80中配混来制备用于配制品xiv的纳米结晶伊曲康唑。然后将129.625g500μm聚苯乙烯研磨介质(密歇根州米德兰的陶氏化学公司)加入悬浮液中,并且将悬浮液以1000rpm研磨30分钟,然后收集。研磨悬浮液的最终中值颗粒大小(dv(50))是258nm。下文将该过程称为“湿磨过程#2”。使用qualificationmicronizer喷射研磨机(美国马萨诸塞州汉诺威市的sturtevant公司(sturtevant,hanover,mausa))制备用于配制品xv的微晶伊曲康唑。将进料压力设定为90psig,并且将研磨压力设定为40psig。将伊曲康唑连续进料到研磨机中,直至研磨60.3g伊曲康唑。研磨api的最终中值颗粒大小(dv(50))是1600nm。下文将该过程称为“喷射研磨过程#1”。然后将用于配制品xv的微粉化伊曲康唑配混到由去离子水中的10wt%伊曲康唑和1.0wt%聚山梨醇酯80组成的悬浮液中。批量大小是200g。通过磁力搅拌棒将聚山梨醇酯80溶解在89.0%di水中,然后缓慢加入伊曲康唑并使其混合直至观察到悬浮液在视觉上分散和均匀。制备原料溶液并将其用于制造由结晶伊曲康唑、聚山梨醇酯80和其他另外的赋形剂组成的干燥粉末。目标是在干基上50wt%伊曲康唑的药物负载。用于喷雾干燥颗粒的原料溶液如下制备。将所需量的水称入适当大小的玻璃容器中。将赋形剂加入水中,并且搅拌溶液直至视觉上澄清。然后将含有伊曲康唑的悬浮液加入赋形剂溶液中并搅拌直至视觉上均匀。然后喷雾干燥原料。在喷雾干燥的同时搅拌原料。原料质量是166.67g至1219.4g,这支持30分钟至3.5小时的制造活动。表27列出了用于制备干燥粉末的原料组分。表27:原料组成用旋风分离器粉末收集,通过在büchib-290minispraydryer(瑞士弗拉维尔的步琪实验室技术服务有限公司)上喷雾干燥由这些原料制造配制品xii-xv的干燥粉末。该系统使用氮气作为干燥和雾化气体以开环(单程)模式运行。液体进料的雾化利用带有1.5mm盖子和0.7液体尖端的büchi喷嘴。调节系统的吸气器以将系统压力维持在-2.0”水柱。遵循以下喷雾干燥条件以制造干燥粉末。对于配制品xii、xiv、和xv,液体原料固体浓度是3%,工艺气体入口温度是127℃至140℃,工艺气体出口温度是60℃,干燥气体流速是17.0kg/hr,雾化气体流速是30.0g/min,并且液体原料流速是6.0ml/min。所得干燥粉末配制品报告于表28中。遵循以下喷雾干燥条件以制造干燥粉末。对于配制品xiii,液体原料固体浓度是3%,工艺气体入口温度是134℃,工艺气体出口温度是60℃,干燥气体流速是17.0kg/hr,雾化气体流速是30.4g/min,并且液体原料流速是6.0ml/min。所得干燥粉末配制品报告于表28中。表28:干燥粉末组成,干基b.粉末表征。四种配制品的本体颗粒大小表征见于表29。配制品xii-xv的1巴下跨度小于2.10,表明相对较窄的大小分布。配制品xii-xv的1巴/4巴分散性比小于1.25,表明它们相对独立于分散能量,这是一种希望的特征,其允许在一系列分散能量内产生类似的颗粒分散。表29:本体颗粒大小测量配制品xii、xiv、和xv以60升/分钟(lpm)和30lpm模拟的患者流速测量和/或计算的几何颗粒大小和胶囊发射的粉末质量(cepm)并报告在表30中。从60lpm至30lpm的cepm和几何大小的微小变化表明干燥粉末配制品相对独立于患者吸入流速,这表明以不同流速呼吸的患者将接受相对类似的治疗剂量。由于缺乏足够的材料量,未对配制品xiii进行发射的颗粒大小测试。表30:发射颗粒大小nt=未测试用八阶段anderson级联冲击器(aci-8)或下一代冲击器(ngi)测量和/或计算的空气动力学颗粒大小、细颗粒分数和细颗粒剂量报告于表31中。配制品xii至xv的细颗粒剂量均表明大于30%的标称剂量到达冲击器阶段,并且因此预测将递送到肺部。配制品xii至xv的mmad在从3.42至4.76范围内,表明在中央和引导气道中沉积。表31:空气动力学颗粒大小通过tga测量配制品vii和viii的重量损失,并且在表32中详述。表32:通过tga的重量损失(%)用hplc-uv测量配制品vii和viii的伊曲康唑含量,并且在表33中详述。表33:伊曲康唑含量通过xrd评估配制品xii的结晶度。在该配制品中观察到伊曲康唑的衍射图案,表明研磨或喷雾干燥过程不影响伊曲康唑的固态。在图案中观察到的另外的峰对应于配制品中的另外的赋形剂。(图6)通过xrd评估配制品xiii的结晶度。在该配制品中观察到伊曲康唑的衍射图案,表明研磨或喷雾干燥过程不影响伊曲康唑的固态。在图案中观察到的另外的峰对应于配制品中的另外的赋形剂。(图7)通过xrd评估配制品xiv的结晶度。在该配制品中观察到伊曲康唑的衍射图案,表明研磨或喷雾干燥过程不影响伊曲康唑的固态。在图案中观察到的另外的峰对应于配制品中的另外的赋形剂。(图8)通过xrd评估配制品xv的结晶度。在该配制品中观察到伊曲康唑的衍射图案,表明研磨或喷雾干燥过程不影响伊曲康唑的固态。在图案中观察到的另外的峰对应于配制品中的另外的赋形剂。(图9)实例8.含有硫酸钠/亮氨酸和水平降低的聚山梨醇酯80的聚山梨醇酯80稳定的结晶伊曲康唑的干燥粉末配制品a.粉末制备。使用qualificationmicronizer喷射研磨机(美国马萨诸塞州汉诺威市的sturtevant公司)制备用于配制品xvi的微晶伊曲康唑。将进料压力设定为90psig,并且将研磨压力设定为40psig。将伊曲康唑(sms制药公司,批号itz-0715005)连续进料到研磨机中,直至研磨约60g伊曲康唑。研磨api的最终中值颗粒大小(dv(50))是约1510nm。然后将用于配制品xvi的微晶伊曲康唑配混到由去离子水中的10wt%伊曲康唑和0.25wt%聚山梨醇酯80组成的悬浮液中。批量大小是440g。通过磁力搅拌棒将聚山梨醇酯80溶解在89.75%di水中,然后缓慢加入微粉化伊曲康唑并使其混合直至观察到悬浮液在视觉上分散和均匀。制备原料溶液并将其用于制造由纳米结晶伊曲康唑、聚山梨醇酯80和其他另外的赋形剂组成的干燥粉末。目标是在干基上50wt%伊曲康唑的药物负载。用于喷雾干燥颗粒的原料溶液如下制备。将所需量的水称入适当大小的玻璃容器中。将赋形剂加入水中,并且搅拌溶液直至视觉上澄清。然后将含有伊曲康唑的悬浮液加入赋形剂溶液中并搅拌直至视觉上均匀。然后将原料喷雾干燥。原料体积是3000g,这支持大约1小时的制造活动。表34列出了用于制备干燥粉末的原料组分。表34:原料组成通过在具有袋式过滤器收集的niromobileminor喷雾干燥器(马里兰州哥伦比亚的gea工程工艺公司)上喷雾干燥,由该原料制造配制品xvi的干燥粉末。该系统使用氮气作为干燥和雾化气体以开环(单程)模式运行。液体进料的雾化利用具有1.0mm液体插入物的schlick940-0雾化器。调节系统的吸气器以将系统压力维持在-2.0”水柱。遵循以下喷雾干燥条件以制造干燥粉末。对于配制品xvi,液体原料固体浓度是1.2%,工艺气体入口温度是181℃至185℃,工艺气体出口温度是65℃,干燥气体流速是80kg/hr,雾化气体流速是250g/min,雾化器入口处的雾化气体背压是30.4psig至31.4psig,并且液体原料流速是50ml/min。所得干燥粉末配制品报告于表35中。表35:干燥粉末组成,干基配制品干燥粉末组成(w/w),干基xvi50%伊曲康唑、35%硫酸钠、13.75%亮氨酸、1.25%聚山梨醇酯80b.粉末表征。该配制品的本体颗粒大小表征见于表36。配制品xvii的1巴下跨度是1.93,表明相对较窄的大小分布。配制品xviii的1巴/4巴分散性比是1.03,表明颗粒大小相对独立于分散能量,这是一种希望的特征,其允许在一系列分散能量内产生类似的分散。表36:本体颗粒大小通过tga测量配制品xvi的重量损失,并且发现为0.37%。通过xrd评估配制品xvi的结晶度。在该配制品中观察到伊曲康唑的衍射图案,表明研磨或喷雾干燥过程不影响伊曲康唑的固态。在图案中观察到的另外的峰对应于配制品中的另外的赋形剂。(图10)实例9.含有硫酸钠/亮氨酸的聚山梨醇酯80稳定的纳米结晶两性霉素b的干燥粉末配制品a.粉末制备。通过在30ml玻璃广口瓶中将4单独等份的1.9g两性霉素b(synbiotics公司15a02no3)在16.96g水和0.190g聚山梨醇酯80(acrosorganics公司,a0365196)中配混来制备纳米结晶两性霉素b。然后将57.88g300μm氧化钇稳定的氧化锆(ytz)陶瓷研磨介质(日本东曹株式会社)加入悬浮液中,并且将悬浮液以200rpm研磨21小时,然后收集。将各个样品合并以制备一批悬浮液研磨悬浮液的最终中值颗粒大小(dv(50))是134nm。制备原料溶液并将其用于制造由纳米结晶两性霉素b、聚山梨醇酯80和其他另外的赋形剂组成的干燥粉末。目标是在干基上50wt%两性霉素b的药物负载。用于喷雾干燥颗粒的原料溶液如下制备。将所需量的水称入适当大小的玻璃容器中。将赋形剂加入水中,并且搅拌溶液直至视觉上澄清。然后将含有两性霉素b的悬浮液加入赋形剂溶液中并搅拌直至视觉上均匀。然后将原料喷雾干燥。原料体积是250g,这支持大约45分钟的制造活动。表37列出了用于制备干燥粉末的原料组分。表37:原料组成用旋风分离器粉末收集,通过在büchib-290minispraydryer(瑞士弗拉维尔的步琪实验室技术服务有限公司)上喷雾干燥由该原料制造配制品xvii的干燥粉末。该系统使用氮气作为干燥和雾化气体以开环(单程)模式运行。液体进料的雾化利用带有1.5mm盖子和0.7mm液体尖端的büchi喷嘴。调节系统的吸气器以将系统压力维持在-2.0”水柱。遵循以下喷雾干燥条件以制造干燥粉末。对于配制品xvii,液体原料固体浓度是2.0%,工艺气体入口温度是132℃至138℃,工艺气体出口温度是60℃,干燥气体流速是17.0kg/hr,雾化气体流速是30.4g/min,并且液体原料流速是6.0ml/min。所得干燥粉末配制品报告于表38中。表38:干燥粉末组成,干基配制品干燥粉末组成(w/w),干基xvii50%两性霉素b、35%硫酸钠、10%亮氨酸、5%聚山梨醇酯80b.粉末表征。该配制品的本体颗粒大小表征见于表39。配制品xvii的1巴下跨度是2.02,表明相对较窄的大小分布。配制品xvii的1巴/4巴分散性比是1.02,表明颗粒大小相对独立于分散能量,这是一种希望的特征,其允许在一系列分散能量内产生类似的分散。表39:本体颗粒大小用下一代冲击器(ngi)测量和/或计算的空气动力学颗粒大小、细颗粒分数和细颗粒剂量报告于表40中。配制品xvii的细颗粒剂量表明高百分比的填充到胶囊中的标称剂量到达冲击器阶段(40.5%),并且因此预测将递送到肺部。配制品xvii的mmad是3.90微米,表明在中央和引导气道中沉积。表40:空气动力学颗粒大小通过tga测量配制品xvii的重量损失,并且发现为3.58%。通过xrd评估配制品xvii的结晶度。在该配制品中观察到两性霉素b的衍射图案,表明研磨或喷雾干燥过程不影响伊曲康唑的固态。在图案中观察到的另外的峰对应于配制品中的另外的赋形剂。(图11)实例10.含有氯化钠/亮氨酸的聚山梨醇酯80稳定的纳米结晶两性霉素b的干燥粉末配制品a.粉末制备。通过在30ml玻璃广口瓶中将4单独等份的1.9g两性霉素b(synbiotics公司15a02no3)在16.96g水和0.190g聚山梨醇酯80(acrosorganics公司a0365196)中配混来制备纳米结晶两性霉素b。然后将57.88g300μm氧化钇稳定的氧化锆(ytz)陶瓷研磨介质(日本东曹株式会社)加入悬浮液中,并且将悬浮液以200rpm研磨21小时,然后收集。将各个样品合并以制备一批悬浮液研磨悬浮液的最终中值颗粒大小(dv(50))是134nm。制备原料溶液并将其用于制造由纳米结晶两性霉素b、聚山梨醇酯80和其他另外的赋形剂组成的干燥粉末。目标是在干基上50wt%两性霉素b的药物负载。用于喷雾干燥颗粒的原料溶液如下制备。将所需量的水称入适当大小的玻璃容器中。将赋形剂加入水中,并且搅拌溶液直至视觉上澄清。然后将含有两性霉素b的悬浮液加入赋形剂溶液中并搅拌直至视觉上均匀。然后将原料喷雾干燥。原料体积是250g,这支持大约45分钟的制造活动。表41列出了用于制备干燥粉末的原料组分。表41:原料组成用旋风分离器粉末收集,通过在büchib-290minispraydryer(瑞士弗拉维尔的步琪实验室技术服务有限公司)上喷雾干燥由该原料制造配制品xvii的干燥粉末。该系统使用氮气作为干燥和雾化气体以开环(单程)模式运行。液体进料的雾化利用带有1.5mm盖子和0.7mm液体尖端的büchi喷嘴。调节系统的吸气器以将系统压力维持在-2.0”水柱。遵循以下喷雾干燥条件以制造干燥粉末。对于配制品xviii,液体原料固体浓度是2.0%,工艺气体入口温度是131℃至132℃,工艺气体出口温度是60℃,干燥气体流速是17.0kg/hr,雾化气体流速是30.4g/min,并且液体原料流速是6.0ml/min。所得干燥粉末配制品报告于表42中。表42:干燥粉末组成,干基配制品干燥粉末组成(w/w),干基xviii50%两性霉素b、35%氯化钠、10%亮氨酸、5%聚山梨醇酯80b.粉末表征。该配制品的本体颗粒大小表征见于表43。配制品xviii的1巴下跨度是2.27,表明相对较窄的大小分布。配制品xviii的1巴/4巴分散性比是1.01,表明颗粒大小相对独立于分散能量,这是一种希望的特征,其允许在一系列分散能量内产生类似的分散。表43:本体颗粒大小用下一代冲击器(ngi)测量和/或计算的空气动力学颗粒大小、细颗粒分数和细颗粒剂量报告于表44中。配制品xviii的细颗粒剂量表明高百分比的填充到胶囊中的标称剂量到达冲击器阶段(47.4%),并且因此预测将递送到肺部。配制品xviii的mmad是3.91微米,表明在中央和引导气道中沉积。表44:空气动力学颗粒大小通过tga测量配制品xviii的重量损失,并且发现为3.35%。通过xrd评估配制品xviii的结晶度。在该配制品中观察到两性霉素b的衍射图案,表明研磨或喷雾干燥过程不影响伊曲康唑的固态。在图案中观察到的另外的峰对应于配制品中的另外的赋形剂。(图12)实例11.喷雾干燥的伊曲康唑、硫酸钠和亮氨酸的干燥粉末配制品a.粉末制备。制备利用水-四氢呋喃(thf)共溶剂体系的原料溶液并将其用于制造由伊曲康唑、硫酸钠和亮氨酸组成的干燥粉末。目标是在干基上50wt%伊曲康唑的药物负载。用于喷雾干燥颗粒的原料溶液如下制备。将所需量的水称入适当大小的玻璃容器中。将赋形剂加入水中,并且搅拌溶液直至视觉上澄清。将所需量的thf称入适当大小的玻璃容器中。将伊曲康唑加入thf中,并且搅拌溶液直至视觉上澄清。然后将含有伊曲康唑的thf溶液加入赋形剂溶液中并搅拌直至视觉上均匀。然后将原料喷雾干燥。原料体积是5l,这支持大约8.5小时的制造活动。表45列出了用于制备干燥粉末的原料组分。表45:原料组成用旋风分离器粉末收集,通过在büchib-290minispraydryer(瑞士弗拉维尔的步琪实验室技术服务有限公司)上喷雾干燥由该原料制造配制品xvii的干燥粉末。该系统使用氮气作为干燥和雾化气体以开环(单程)模式运行。液体进料的雾化利用带有1.5mm盖子和0.7mm液体尖端的büchi喷嘴。调节系统的吸气器以将系统压力维持在-2.0”水柱。遵循以下喷雾干燥条件以制造干燥粉末。对于配制品xix,液体原料固体浓度是12.0g/l,工艺气体入口温度是92℃至103℃,工艺气体出口温度是40℃,干燥气体流速是17.0kg/hr,雾化气体流速是2830g/min,并且液体原料流速是10.0ml/min。所得干燥粉末配制品报告于表46中。表46:干燥粉末组成,干基配制品干燥粉末组成(w/w),干基xix50%伊曲康唑、35%硫酸钠、15%亮氨酸b.粉末表征。该配制品的本体颗粒大小表征见于表47。配制品xix的1巴下跨度是2.32,表明相对较窄的大小分布。配制品xix的1巴/4巴分散性比是1.12,表明颗粒大小相对独立于分散能量,这是一种希望的特征,其允许在一系列分散能量内产生类似的分散。表47:本体颗粒大小测量该配制品以60升/分钟(lpm)和30lpm模拟的患者流速测量和/或计算的几何颗粒大小和胶囊发射的粉末质量(cepm)并报告在表48中。从60lpm至20lpm的cepm和几何大小的微小变化表明干燥粉末配制品相对独立于患者吸入流速,这表明以不同流速呼吸的患者将接受相对类似的治疗剂量。表48:发射颗粒大小用下一代冲击器(ngi)测量和/或计算的空气动力学颗粒大小、细颗粒分数和细颗粒剂量报告于表49中。配制品xix的细颗粒剂量表明高百分比的填充到胶囊中的标称剂量到达冲击器阶段(41.1%),并且因此预测将递送到肺部。配制品xix的mmad是3.80微米,表明在中央和引导气道中沉积。表49:空气动力学颗粒大小通过tga测量配制品xix的重量损失,并且发现为0.37%。用hplc-uv测量配制品xix的伊曲康唑含量并且是99.0%。通过xrd评估配制品xix的结晶度(图13)。未观察到伊曲康唑峰,表明配制品中不存在明显水平的伊曲康唑。如图所示,在配制品中观察到的所有峰均对应于赋形剂。因此,配制品xix中伊曲康唑的固态可以表征为无定形。实例12.含有伊曲康唑的干燥粉末配制品的体外溶解研究a.体外溶解研究利用体外模型来提供预测测试以了解伊曲康唑的溶解。药物溶解是细胞通过肺部摄取和/或吸收的先决条件。因此,伊曲康唑的溶解动力学在确定其从呼吸道吸收的程度方面起关键作用。对于含有作为气溶胶被递送到呼吸道的伊曲康唑的干燥颗粒,伊曲康唑在这些颗粒中的命运取决于它们的物理化学特性。对于雾化干燥颗粒中的伊曲康唑在肺中发挥局部作用,干燥颗粒必须首先经历溶解以使伊曲康唑存在于肺液和组织中,从而作用于真菌感染。然而,一旦发生伊曲康唑溶解到肺液中,则伊曲康唑可以进一步用于渗透和全身吸收。预测伊曲康唑的溶解速率与其溶解度、周围液膜中的浓度和固液界面面积成正比。溶解度取决于药物的化合物、配制品和物理形式。肺中的总液体体积是10-30ml,其中内液体积对应于ca.5μl/cm2,这可能损害难溶性分子(如伊曲康唑)的溶解和随后的吸收。将以下体外溶解模型用于理解含有伊曲康唑的干燥粉末气溶胶的溶解特性。使用下一代冲击器(ngi)(英国的科普利科技有限公司)以明确定义的气溶胶颗粒大小分布(apsd)截止值收集气溶胶颗粒,并且然后使用模型肺液模拟溶解行为。将unidose(tm)(英国新港的nanopharm公司(nanopharm,newport,unitedkingdom))气溶胶剂量收集系统与改进的下一代冲击器(ngi)组合用于将冲击器阶段质量(ism)(其定义为在下一代冲击器的阶段2上和以下收集的剂量)均匀沉积在适合过滤器上用于在uspv-桨碟法(pod)装置中进行随后的溶解研究。b.用于体外溶解研究的材料和方法本研究中所使用的材料在表50中示出。将粉末配制品、胶囊和包装材料在22.5℃±2.5℃和30±5%rh下平衡。在相同条件下将配制品包封在3号hpmc胶囊中。粉末制剂的填充重量是10mg。在单位剂量的基于胶囊的dpi装置(rs01,意大利osnago的plastiape公司)中从胶囊雾化配制品。使用plastiapers01干燥粉末吸入器(dpi)以60l/min(4l吸入体积)雾化每种配制品的一个胶囊。在unidose系统中收集气溶胶剂量。使用micromisttm雾化器(美国加利福尼亚州特曼库拉的hudsonrci公司(hudsonrci,temecula,ca,usa))以15l/min将1毫升悬浮液配制品雾化成cngi。将unidose收集系统用于使整个冲击器阶段质量(即,ngi的阶段2以下)均匀地沉积到玻璃微纤维滤膜上,其可以被看作圆圈(代表颗粒或液滴)沉积的位置。将过滤器置于盘盒中,并在usp装置iipod(桨碟法,uspv)中于37℃下使用500mlpbsph7.4+2.0%sds进行溶解研究。对于所有研究,在容器内保持沉降条件。在指定的时间点取样并在agilent(美国加利福尼亚州圣克拉拉)1260infinity系列hplc上测试药物含量。数据以240分钟(min)的原始累积质量和累积质量百分比(%)呈现。表50.测试的配制品。c.配制品的冲击器阶段质量(ism)的unidosepod溶解研究结果。配制品的ism的原始累积质量和百分比累积质量溶解曲线分别在图14和15中示出。每种粉末配制品的unidoseism和溶解半衰期总结在表50中。悬浮液中的伊曲康唑晶体的颗粒大小和使用测量的颗粒大小分布估计的伊曲康唑晶体的比表面积(ssa)也在表51中示出。基于累积质量数据,所收集的配制品的ism范围在2.1-2.6mg伊曲康唑之间。这些数据表明,基于标称剂量,配制品的雾化效率是大约50%,因为每个颗粒中的伊曲康唑负载是50%并且标称剂量是5mg伊曲康唑(10mg粉末)。配制品xix的溶解速率最快,并且多于80%的药物在第一个时间点内溶解。由于配制品xix的快速溶解动力学,因此不可能计算溶解半衰期。其他粉末配制品的溶解半衰期在其溶解动力学中显示以下等级顺序:xi>xii>xiii>xiv>xv>纯itz还评估了图14和15中所示数据的配制品xi、xii、xiii、xiv、和xv的颗粒大小与它们各自的溶解半衰期之间的关系,如图16a所示。这些数据表明伊曲康唑晶体的颗粒大小与溶解半衰期之间的良好相关性。图16b示出了伊曲康唑晶体的比表面积与溶解半衰期之间的关系。这些数据表明,随着配制品中颗粒的表面积增加,溶解半衰期缩短。这些数据强调了药物的颗粒大小和因此表面积影响配制品的溶解行为。基于实例14中示出的药代动力学数据,配制品xix具有最高的全身暴露。这与溶解数据相关,这表明该配制品具有快速溶解动力学。表示为配制品xix的cmax响应比率的其他粉末配制品的溶解半衰期与cmax之间的关系或cmax,分别在图17和18中示出。溶解半衰期与cmax之间存在相反关系,这表明更快的溶解速率导致更高的全身暴露。配制品xix的全身响应的cmax比率与溶解半衰期之间的相关性更强。这些数据表明,伊曲康唑配制品的全身暴露响应由其溶解行为并且继而由配制品的物理化学特性来调整。表51.每种配制品的颗粒大小、unidoseism和溶解半衰期在以下列出。由unidosepod测定的纳米悬浮液配制品和微悬浮液配制品的ism的原始累积质量百分比溶解曲线在图19中示出。纳米悬浮液配制品的溶解速率比微悬浮液配制品更快。纳米悬浮液配制品和微悬浮液配制品的溶解半衰期分别是5.3和35.5min。实例13.含有结晶伊曲康唑的干燥粉末配制品的体外溶解和渗透性研究。a.体外溶解和渗透性研究基于使用基于细胞的体外方法模拟呼吸上皮表面处的气液界面,使用生物相关溶解测试系统。将使细胞培养板并入收集阶段(cngi)上的改良的下一代冲击器用于将材料均匀地沉积到细胞培养物上。测量药物通过上皮细胞单层的溶解和渗透。b.用于体外溶解和渗透性研究的材料和方法将在snapwelltm(美国马萨诸塞州的corningcostar公司(corningcostar,massachusetts,usa))可渗透插入物中的气-液界面处生长的上皮细胞单层整合到cngi中。使calu-3细胞系(atcc,英国泰丁敦的lgc标准品公司(lgcstandards,teddington,uk))(第32-50代)在补充有非必需氨基酸、10%(v/v)胎牛血清、1%(v/v)青霉素-链霉素和1%(v/v)两性霉素抗真菌剂并分别在37℃下在95%/5%空气/co2的潮湿气氛中维持的最小必需培养基(mem)中生长。将细胞以5×105个细胞.cm-2的密度接种到snapwell插入物上,并在培养第2天的空气界面条件下培养12天。使用连接到evom2epithelialvoltohmmeter(英国希钦的世界精密仪器公司(worldprecisioninstruments,hitchin,unitedkingdom))的evom2筷子电极测量跨上皮电阻(teer),并且认为teer高于450ω.cm2的单层是汇合的。将含有calu-3ali细胞的snapwell转移到改进的ngi杯并置于ngi(英国诺丁汉的科普利科技有限公司)的第4阶段中。将粉末配制品的单个胶囊以60l/min持续4秒雾化到cngi中。使用micromist雾化器(美国加利福尼亚州特曼库拉的hudsonrci公司)以15l/min将1毫升悬浮液配制品雾化成cngi。本研究中所使用的材料在表49中示出。将粉末配制品、胶囊和包装材料在22.5℃±2.5℃和30±5%rh下平衡。在相同条件下将配制品包封在3号hpmc胶囊中。粉末制剂的填充重量是10mg。在单位剂量的基于胶囊的dpi装置(rs01,意大利osnago的plastiape公司)中从胶囊雾化配制品。使用plastiapers01干燥粉末吸入器(dpi)以60l/min(4l吸入体积)雾化每种配制品的一个胶囊。在将剂量给药至第4阶段的snapwell后,将snapwell转移到6孔板,其含有保持在37℃下的2mlpbsph7.4+2.0%sds。在不同的时间点在基底外侧取样并在agilent(美国加利福尼亚州圣克拉拉)1260infinity系列hplc上测量药物含量。递送到细胞的总剂量由在时间过程中溶解的药物总量和实验后裂解细胞的总量来测量。c.伊曲康唑粉末配制品的cngi综合溶解和渗透性研究结果图20中示出了在阶段4递送到细胞的伊曲康唑粉末配制品的总回收剂量图的累积质量百分比(%)。这些数据表明不同配制品的溶解与渗透性动力学之间的差异。未作改变的纯itz比其他配制品具有更慢的溶解和渗透性动力学,而配制品xix具有最快的溶解和渗透性动力学。为了理解不同配制品的cngi数据,我们通过考虑负载剂量差异利用该数据来计算药物物质的扩散速率。这是使用以下方程完成的:其中j是通量(cngi溶解/渗透性曲线的梯度),a是屏障的面积,并且c0是负载剂量。这些数据总结在表52中,显示配制品的扩散速率遵循以下等级顺序:xix>xi>xii>xiii>xiv>xv>纯itz表52.以下每种粉末配制品的颗粒大小和扩散速率。基于实例14中示出的药代动力学数据,配制品xix具有最高的全身暴露。这与该配制品的扩散速率相关,这表明该配制品具有快速溶解和渗透动力学。表示为配制品xix的cmax响应比率的其他粉末配制品的扩散速率与cmax之间的关系或cmax,分别在图21和22中示出。扩散速率与cmax之间存在关系,这表明更快的扩散速率导致更高的全身暴露。配制品xix的全身响应的cmax比率与扩散速率之间的相关性更强。由cngi测定的纳米悬浮液配制品和微悬浮液配制品的ism的原始累积质量百分比溶解曲线在图23中示出。cngi数据表明,纳米悬浮液配制品的扩散速率比微悬浮液配制品更快。实例14.大鼠中的单剂量吸入pk研究a.材料和方法在-60分钟的暴露期间内单次吸入给予五种不同的伊曲康唑配制品中的每一种后,从大鼠取血和肺组织样品,以便评估雄性大鼠对伊曲康唑及其代谢产物羟基-伊曲康唑的全身暴露,标称剂量水平为5mg/kg。通过验证的lc-ms/ms方法测量在暴露期结束时和暴露结束后长达96小时采集样品中的伊曲康唑和羟基-伊曲康唑的血浆浓度。b.结果–血浆伊曲康唑的最大平均血浆浓度(cmax)和直至最后一个可定量样品时间估计的平均血浆浓度-时间曲线下面积(auc最后)总结在表53中。表53.血浆cmax和auc最后基于校正了所接收剂量的差异的cmax和auc最后值,每组中最大平均血浆浓度(cmax)和平均血浆浓度-时间曲线下面积(auc最后)相对于接受配制品xix的组的cmax和auc最后值的比率呈现于表54中。表54.血浆cmax和auc最后,二者均相对于配制品xix。在暴露于配制品xix后,大鼠全身暴露于伊曲康唑的速率(cmax)和程度(auc最后)最高。在暴露于配制品xii和配制品xi后,cmax和auc最后是相似的,并且在暴露于配制品xiv后略低。在暴露于配制品xv后,cmax和auc最后最低。观察到全身暴露于羟基-伊曲康唑的速率和程度的类似模式,尽管暴露于配制品xii后的cmax和auc最后值低于暴露于配制品xi后的cmax和auc最后值,并且略高于暴露于配制品xiv后的cmax和auc最后值。c.结果–肺组织伊曲康唑的最大平均肺组织浓度(cmax)和直至最后一个可定量样品时间估计的平均肺组织浓度-时间曲线下面积(auc最后)总结在表55中。表55.肺组织cmax和auc最后基于校正了所接收剂量的差异的cmax和auc最后值,每组中最大平均肺组织浓度(cmax)和平均肺组织浓度-时间曲线下面积(auc最后)相对于接受配制品xix的组的cmax和auc最后值的比率呈现于表56中。表56.肺组织cmax和auc最后,二者均相对于配制品xix。在暴露于配制品xix后,大鼠肺部局部暴露于伊曲康唑的速率(cmax)和程度(auc最后)最低。在暴露于配制品xii、配制品xi和配制品xiv后,cmax和auc最后通常相似,尽管暴露于配制品xii后的auc最后略低于暴露于其他两种配制品后的auc最后。在暴露于配制品xv后,cmax仅略高于暴露于配制品xix后的cmax并且低于其他配制品的值,而auc最后高于暴露于配制品xix后的auc最后,并且与暴露于其他配制品后的auc最后大致相似。羟基-伊曲康唑的cmax和auc最后值在暴露于配制品xix后最高,并且在暴露于配制品xii、配制品xi、配制品xiv和配制品xv后较低,但对于所有这四种配制品大致相似。肺中auc最后值与血浆中相应值的比率呈现于表57中。表57.肺组织与血浆组织的auc最后比率伊曲康唑的肺组织:血浆比率在暴露于配制品xix后最低,在暴露于配制品xii和配制品xi后相似,并且在暴露于配制品xiv后略高。在暴露于配制品xv后观察到最高比率。羟基-伊曲康唑的肺组织:血浆比率在暴露于每种配制品后相似,并且远低于对伊曲康唑观察到的比率。结论在给予配制品xix后,大鼠对伊曲康唑的全身暴露最高。在吸入给予配制品xii和配制品xi后的全身暴露是相似的,并且在给予配制品xiv后略低。在给予配制品xv后的全身暴露最低。对于全身暴露于羟基-伊曲康唑观察到相似的模式,在给予配制品xii后的全身暴露低于给予配制品xi后的全身暴露,并且略高于给予配制品xiv后的全身暴露。在暴露于配制品xix后,大鼠肺部对伊曲康唑的局部暴露最低。在给予配制品xii、配制品xi和配制品xiv后的局部暴露通常相似。在给予配制品xv后,最大浓度仅略高于给予配制品xix后的最大浓度,并且低于其他配制品的值,而auc最后值高于暴露于配制品xix后的auc最后值,并且与暴露于其他配制品后的auc最后值大致相似。在给予配制品xix后局部暴露于羟基-伊曲康唑最高,并且在给予配制品xii、配制品xi、配制品xiv和配制品xv后较低,但对于所有这四种配制品大致相似。实例15.制备用于28天毒性研究的无定形伊曲康唑的干燥粉末配制品a.粉末制备。制备利用水-四氢呋喃(thf)共溶剂体系的原料溶液并将其用于制造由伊曲康唑、硫酸钠和亮氨酸组成的干燥粉末。目标是在干基上50wt%伊曲康唑的药物负载。用于喷雾干燥颗粒的原料溶液如下制备。将所需量的水称入适当大小的玻璃容器中。将赋形剂加入水中,并且搅拌溶液直至视觉上澄清。将所需量的thf称入适当大小的玻璃容器中。将伊曲康唑加入thf中,并且搅拌溶液直至视觉上澄清。然后将含有伊曲康唑的thf溶液加入赋形剂溶液中并搅拌直至视觉上均匀。然后将原料喷雾干燥。单个原料体积是9.5625l。制备了这些原料中的14种,总共133.875l,这支持了大约30小时的制造活动。表58列出了用于制备干燥粉末的每种原料组分。表58:原料组成通过在具有袋式过滤器收集的niromobileminor喷雾干燥器(马里兰州哥伦比亚的gea工程工艺公司)上喷雾干燥,由该原料制造配制品xx的干燥粉末。该系统使用氮气作为干燥和雾化气体以开环(单程)模式运行。液体进料的雾化利用具有1.0mm液体插入物的niro雾化器。调节系统的吸气器以将系统压力维持在-2.0”水柱。遵循以下喷雾干燥条件以制造干燥粉末。对于配制品xx,液体原料固体浓度是12g/l,工艺气体入口温度是120℃至140℃,工艺气体出口温度是40℃,干燥气体流速是80kg/hr,雾化气体流速是352.2g/min,雾化器入口处的雾化气体背压是45psig至57psig,并且液体原料流速是75ml/min。所得干燥粉末配制品报告于表59中。配制品中的伊曲康唑是无定形的。表59:干燥粉末组成,干基配制品干燥粉末组成(w/w),干基xx50%伊曲康唑、35%硫酸钠、15%亮氨酸b.粉末表征。该配制品的本体颗粒大小表征见于表60。配制品xx的1巴下跨度是1.83,表明相对较窄的大小分布。配制品xx的1巴/4巴分散性比是1.06,表明颗粒大小相对独立于分散能量,这是一种希望的特征,其允许在一系列分散能量内产生类似的分散。表60:本体颗粒大小通过tga测量配制品xx的重量损失,并且发现为0.34%。用hplc-uv测量配制品xx的伊曲康唑含量并且是标称的100.9%。实例16.制备用于28天毒性研究的结晶伊曲康唑的干燥粉末配制品a.粉末制备。将用于配制品xxi的纳米结晶伊曲康唑制备为包含25wt%伊曲康唑(sms制药公司批号itz-0715005)和2.5wt%聚山梨醇酯的悬浮液。通过磁力搅拌棒将聚山梨醇酯80溶解在72.5%去离子水中,然后加入伊曲康唑并通过用磁力搅拌棒搅拌悬浮。一旦所有伊曲康唑悬浮,则使用0.2mm研磨介质(日本东京的东曹株式会社(tosoh,tokyo,japan))在netzschminicer上处理配制品,其中室填充率为90%。将以下条件用于制造伊曲康唑悬浮液。研磨机速度是3000rpm,进口泵速度是100rpm,循环冷却器是10℃,入口空气压力是4.5巴,并且运行时间是30-40分钟。以这种方式处理八种悬浮液并合并以制成最终的悬浮液批次。研磨悬浮液的最终中值颗粒大小(dv(50))是130nm。将用于配制品xxii的纳米结晶伊曲康唑制备为在去离子水中包含10wt%伊曲康唑和0.7wt%油酸、1.5%氢氧化铵的悬浮液。通过磁力搅拌棒将油酸溶解在87.8去离子水中,并且然后加入氢氧化铵并通过磁力搅拌棒溶解。最后,加入伊曲康唑并用磁力搅拌棒混合以形成悬浮液。一旦所有伊曲康唑悬浮,则使用0.5mm研磨介质(日本东京的东曹株式会社)在netzschminicer上处理配制品,其中室填充率为90%。将以下条件用于制造伊曲康唑悬浮液。研磨机速度是3000rpm,进口泵速度是100rpm,循环冷却器是10℃,入口空气压力是4.5巴,并且运行时间是200-240分钟。以这种方式处理八种悬浮液并合并以制成最终的悬浮液批次。研磨悬浮液的最终中值颗粒大小(dv(50))是115nm。使用qualificationmicronizer喷射研磨机(美国马萨诸塞州汉诺威市的sturtevant公司)制备用于配制品xxiii的微晶伊曲康唑。将进料压力设定为85psig,并且将研磨压力设定为45psig。将伊曲康唑连续进料到研磨机中,直至研磨480.0g伊曲康唑。研磨api的最终中值颗粒大小(dv(50))是1640nm。然后将用于配制品xxiii的微粉化伊曲康唑配混到由去离子水中的10wt%伊曲康唑和0.25wt%聚山梨醇酯80组成的悬浮液中。批量大小是4800g。通过磁力搅拌棒将聚山梨醇酯80溶解在88.75%去离子水中,然后缓慢加入伊曲康唑并使其混合直至观察到悬浮液在视觉上分散和均匀。制备原料悬浮液并将其用于制造由结晶伊曲康唑和其他另外的赋形剂组成的干燥粉末。目标是在干基上50wt%伊曲康唑的药物负载。用于喷雾干燥颗粒的原料悬浮液如下制备。将所需量的水称入适当大小的玻璃容器中。将赋形剂加入水中,并且搅拌溶液直至视觉上澄清。然后将含有伊曲康唑的悬浮液加入赋形剂溶液中并搅拌直至视觉上均匀。然后喷雾干燥原料。在喷雾干燥的同时搅拌原料。用于配制品xxi的单个原料质量各为7.5kg。喷雾干燥这些原料中的六种,这支持了十五小时的制造活动。用于配制品xxii的单个原料质量各为6.0kg。喷雾干燥这些原料中的三种,这支持了六小时的制造活动。用于配制品xxiii的单个原料质量各为8.0kg。喷雾干燥这些原料中的四种,这支持了大约11小时的制造活动。表61列出了用于制备干燥粉末的原料组分。表61:含有聚山梨醇酯80的配制品的原料组成表62:含有油酸的配制品的原料组成通过在具有袋式过滤器收集的niromobileminor喷雾干燥器(马里兰州哥伦比亚的gea工程工艺公司)上喷雾干燥,由这些原料制造配制品xxi-xxiii的干燥粉末。该系统使用氮气作为干燥和雾化气体以开环(单程)模式运行。液体进料的雾化利用具有1.0mm液体插入物的niro双流体喷嘴雾化器。调节系统的吸气器以将系统压力维持在-2.0”水柱。遵循以下喷雾干燥条件以制造干燥粉末。对于配制品xxi和xxii,液体原料固体浓度是3%,工艺气体入口温度是170℃至190℃,工艺气体出口温度是65℃,干燥气体流速是80.0kg/hr,雾化气体流速是250.0g/min,并且液体原料流速是50.0ml/min。所得干燥粉末配制品报告于表64中。对于配制品xxiii,液体原料固体浓度是1.2%,工艺气体入口温度是170℃-190℃,工艺气体出口温度是65℃,干燥气体流速是80.0kg/hr,雾化气体流速是250.0g/min,并且液体原料流速是50.0ml/min。所得干燥粉末配制品报告于表63中。表63:干燥粉末组成,干基b.粉末表征。三种配制品的本体颗粒大小表征见于表64。配制品xxi-xxiii的1巴下跨度小于2.05,表明相对较窄的大小分布。配制品xxi-xxiii的1巴/4巴分散性比小于1.25,表明它们相对独立于分散能量,这是一种希望的特征,其允许在一系列分散能量内产生类似的颗粒分散。表64:本体颗粒大小通过tga测量配制品xxii-xxiii的重量损失,并且在表65中详述。表65:通过tga的重量损失(%)用hplc-uv测量配制品xxi-xxiii的伊曲康唑含量,并且在表66中详述。表66:伊曲康唑含量实例17.大鼠的28天吸入毒性研究a和ba.材料和方法为了评估血浆和肺二者的药代动力学以及局部组织毒性的可能性,进行了两项单独的28天研究。在第一项研究(28天研究a)中,用空气或安慰剂对照或三个剂量的配制品xx中的一种将5组动物每天给药28天。在第二项研究(28天研究b)中,用三种结晶纳米微粒伊曲康唑配制品中的一种将7组大鼠每天给药28天,或者在一组的情况下,每三天给药。组和实现的剂量在表67和68中详述。表67,28天研究a中的剂量组表68,28天研究b中的剂量组在两项研究中,在每种配制品的第一次和最后一次吸入给予后从大鼠取血和肺组织样品,以便评估雄性和雌性大鼠中肺和全身暴露和伊曲康唑的积累。此外,在最后一次给药后24小时从所有动物收集肺组织样品,包括喉、气管、气管分叉(隆突)和肺,以便评估由给药引起的微观病理学变化。通过验证的lc-ms/ms方法测量样品中伊曲康唑的血浆和肺浓度。b.结果–血浆在来自28天研究a(使用无定形伊曲康唑)的雄性和雌性大鼠中第1和第28天的伊曲康唑的最大平均血浆浓度(cmax)和直至最后一个可定量样品时间估计的平均血浆浓度-时间曲线下面积(auc0-最后)总结在表69中,并且来自28天研究b(使用结晶伊曲康唑)的总结在表70中。表69.伊曲康唑的血浆cmax和auc0-最后表70.伊曲康唑的血浆cmax和auc0-最后*每三天给药尽管各种配制品之间绝对实现的剂量存在差异,但很明显pur1920的峰值(cmax)和总体(auc0-最后)全身暴露高于任何结晶配制品,二者均在单剂量(第1天)和重复给药(第28天)后。下表71总结了来自每次研究的使用15mg/kg/天的目标剂量的28天研究的每种配制品的伊曲康唑的剂量归一化平均cmax和auc0-最后。对于每天的每次研究通过将测量的暴露除以实际实现剂量来实现归一化。表71.每种结晶配制品的剂量归一化血浆cmax和auc0-最后。在第1天大鼠中伊曲康唑全身暴露的剂量归一化cmax和auc0-最后在暴露于配制品xx后最高。到第28天,配制品xx通常仍显示相对于结晶配制品xxi更高的剂量归一化cmax和auc0-最后,特别是在雌性中,尽管差异不太明显,其中对于一些配制品雄性中auc0-最后的值略高。这些数据表明,对于给定的实现的递送肺剂量,由吸入结晶配制品引起的全身暴露通常小于配制品xx的全身暴露。然而,鉴于全身暴露依赖于材料在肺中的溶解速率和肺组织的渗透性二者,它还表明,随着时间的推移,结晶配制品确实显示出充分溶解和组织的渗透以缩小或消除差异,从而提供了结晶配制品不仅仅是肺中不溶性沉积物的信心。c.结果–肺组织表示为与相同时间点处相应平均血浆浓度值的比率的每组中第1天和第28天谷平均肺组织浓度(前一剂量结束后23小时)呈现于表72中。表72.每种配制品的肺:血浆浓度比率。在暴露于配制品xx后,伊曲康唑的肺组织:血浆比率最低,并且在第1天和第28天始终比所有结晶配制品高得多。这些数据表明,结晶配制品提供了显著更高的肺暴露,其中在所测试的剂量下具有更少的全身暴露,从而增加了作用部位处的暴露,同时使全身暴露的不希望影响的可能性最小化。d.结果-肺病理学在28天研究a中,配制品xx相关的显微镜检查结果存在于≥5mg/kg/天的呼吸组织中。在所有剂量下均存在最小至轻微的肉芽肿性炎症,并且巨噬细胞和多核巨细胞经常包含胞质内针状体。在其中包括28天恢复期的最高剂量下,这些仅部分恢复。记录的病理学在所有剂量下均被认为是不利的,因为它的分散呈现以及在恢复期间它确实没有完全消退的事实。在病理学中提到的针状形成似乎是伊曲康唑,理论上我们认为其在无定形材料使内液和间质空间过饱和导致多次剂量后api结晶时形成。使用相同配制品的较短持续时间暴露研究显示没有此类发现。在28天研究b中,配制品xxi和xxiii与仅在40mg/kg/天的肺中泡沫巨噬细胞的最小不利累积相关,其中配制品xxi是以该水平给药的唯一配制品。以相当剂量水平给药配制品xxi-xxiii的大鼠之间发现的发病率和严重性没有明显差异。总体而言,对于所有三种测试的结晶配制品,未观察到不良反应水平(noael)是大约15mg/kg/天。与大鼠呼吸道中的无定形组合物相关的病理学发现具有与由结晶配制品诱导的那些不同的特征,其中后一组中的发现对于粘膜内的肉芽肿性炎症更多地与对气道腔中累积材料的清除响应相关。此外,无定形配制品相关的发现涉及呼吸道中的更多区域,并且在较低剂量下是不利的。结论在给予配制品xx后,大鼠对伊曲康唑的全身暴露(即血浆水平)最高。在吸入给予配制品xxi-xxiii后,全身暴露通常较少,尽管在给药的第28天,差异小于单剂量后的差异。然而,相对于配制品xx,使用配制品xxi-xxiii的肺暴露显著且持续地更高。当比较肺和全身暴露时,配制品xx的比率有利于肺暴露超过全身暴露。然而,对于每种结晶配制品xxi-xxiii,肺:血浆比率显著更大。这些数据表明,结晶配制品提供了显著更高的伊曲康唑局部浓度,同时导致与配制品xx相同或更少的全身暴露。配制品xx中伊曲康唑的无定形性质导致溶解度增加和通过肺快速转运到全身循环,如第1天显著更高的全身暴露所证明。配制品xx给药还导致粘膜中呈针状沉积物形式的局部毒性,从而导致肉芽肿性炎症,其在所有测试剂量下均是不利的,并且低至5mg/kg/天。通过在配制品xxi-xxiii中使用结晶纳米颗粒,肺保留显著更大,从而导致比无定形配制品更高的局部暴露,通常具有相同或更少的全身暴露。暴露曲线的这种变化具有增加肺部功效的优点,其中全身性伊曲康唑暴露的不良作用相对于配制品xx没有更差并且可能进一步最小化。此外,尽管局部暴露显著更高,但配制品xxi-xxiii显示出低得多的不良微观病理学发现的可能性。总结体外和体内实例总结对干燥粉末配制品中伊曲康唑物理形式影响的研究涉及通过体外溶解和渗透性研究以及体内单剂量和多剂量药代动力学和毒性研究的迭代进展。体外溶解研究表明,伊曲康唑的物理形式以及配制品中结晶颗粒的大小在确定溶解速率以及预期所递送的材料通过肺并进入全身循环中的速率方面起重要作用。这些数据证明了控制肺和全身暴露二者的关键方面,以允许调整功效以及可能调整不良发现的能力。将这些体外研究结果在体内单剂量吸入pk研究中进行了测试,证实了当通过吸入递送时,相对于含有无定形伊曲康唑的配制品,含有结晶伊曲康唑纳米颗粒的粉末在单剂量后导致更长的肺保留,从而导致更高的肺与血浆比率,以及降低的峰值和总全身暴露。总结28天吸入毒性研究的实例进一步证明,在肺和全身暴露方面,干燥粉末配制品中无定形和结晶伊曲康唑的不同暴露动力学在给药的多天内保持不变。此外,当在多天给药后检查无定形和结晶材料的微观病理学效应时,很明显这些结果的性质和严重性二者中存在差异,其中结晶材料显示较少的不良发现并且仅在较高的肺部暴露下。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1