一种具有化疗光疗联合作用的多功能靶向给药系统及其制备方法与流程
本发明涉及材料制备和药物控制释放技术领域,尤其涉及一种具有化疗光疗联合作用的多功能靶向给药系统及其制备方法。
背景技术:
癌症也称恶性肿瘤,是由控制细胞生长增殖机制失常而引起的疾病。癌症在世界范围内都是一个重大的问题,全球癌症相关的负担也在不断的增加。目前化疗仍然是癌症治疗的主要手段,化疗药物药力强大、可快速杀伤或杀死肿瘤细胞,在临床治疗中发挥着重要作用。但化疗药物普遍存在细胞选择性差、毒副作用大、不良反应严重和多药耐药等问题,使药物难以发挥其最佳的疗效。因此,针对肿瘤组织进行靶向设计和开发新的药物输送体系成为癌症治疗药物的重要研究内容。
光疗方式由于其高效性,非侵入性和远程控制能力,引起了广泛的研究和关注。光疗分为光热疗法和光动力疗法两种方式。光热疗法(photothermaltherapy,ptt)主要依赖于光热剂(photother是malagents,pta)的局部加热效应。光热剂在肿瘤部位聚集后在nir激光照射下,可以将光能转换成局部热量以产生热疗从而破坏肿瘤。光动力疗法(photodynamictherapy,pdt)是近20年内发展起来的一种新型恶性肿瘤治疗方法。利用外加光敏剂或光敏剂前体,增加肿瘤内光敏剂的聚集,随后给予特定波长的激发光激活光敏剂,从而引发细胞氧化应激反应导致肿瘤细胞的死亡。与传统的手术、放疗、化疗相比,光疗具有相对的选择特异性,毒副作用小,非侵入性,无累加毒性等特点,在肿瘤治疗中具有良好的应用前景。
聚多巴胺(polydopamine,pda)由其单体多巴胺(da)的自动氧化聚合产生,具有良好的生物相容性、水溶性和光热特性,可用于肿瘤的光热治疗。在中性或弱碱性介质中,多巴胺可以在有机或无机材料表面上自聚合,这与儿茶酚氧化成醌有关,还可以进一步与胺和其他儿茶酚/醌反应形成粘附聚合物涂层。已有研究表明,多巴胺对基体材料表面的附着行为来源于多巴胺的邻苯二酚和氨基官能团,这种结构可以和有机-无机表面建立共价和非共价的相互作用,从而使聚多巴胺交联层强力附着在材料表面上。且聚多巴胺对ph敏感,表面聚多巴胺修饰的纳米粒子在血液和组织液等中性ph条件下可保持稳定,但在肿瘤部位的酸性条件下会降解并释放药物。
二氢卟吩(chlorine6,ce6)是一种光敏剂,可以通过近红外nir光活化,相对快速地从体内消除并产生单线态氧,从而被广泛用于光动力治疗(pdt)。常用的光敏剂多为水不溶性分子(如:ce6),易团聚,且对肿瘤部位缺乏靶向性,在体内难以递送至肿瘤部位,并富集达到有效浓度,从而影响治疗效果。有研究者将阿霉素和光敏剂二氢卟吩(ce6)共同负载于荧光上转换纳米粒用于杀伤癌细胞,结果表明化疗药物与光敏剂共传递系统的癌细胞杀伤效果明显提高,协同效应显著。
透明质酸(hyaluronicacid,ha)是一种天然的多糖大分子,由d-葡萄糖醛和n-乙酰氨基葡萄糖交替的二糖单元组成,广泛分布在人体组织中,是细胞外基质的重要组成,故不会发生透明质酸分子自身的免疫排斥。天然透明质酸是水溶性高分子,在酶的作用下在体内可以天然降解,因此被广泛用作药物载体。透明质酸可以通过交联、酯化和复合等方法进行修饰之后,再将其与药物进行结合,在透明质酸被降解或被细胞摄入时释放出主要药物,从而达到缓释和控释的作用。cd44是一种跨膜受体糖蛋白,透明质酸与其特异性的相互作用已作为透明质酸靶向治疗恶性肿瘤的研究基础,cd44在多种恶性瘤细胞表面都有过度表达,所以利用透明质酸作为载体,将小分子药物搭载其上,可以在增强药物选择性的同时降低药物对正常细胞的杀伤作用,并可提高药物的溶解性和稳定性。中国发明专利(申请号201510662183.8,申请日2015.10.14)公开了一种邻苯二酚修饰的透明质酸的载药系统及其制备方法,邻苯二酚修饰的透明质酸具有较高的细胞黏付性,使负载阿霉素的邻苯二酚修饰的透明质酸纳米纤维对肿瘤细胞具有更好的治疗作用。
技术实现要素:
基于以上现有技术的不足,本发明所解决的技术问题在于提供一种具有化疗光疗联合作用的多功能靶向给药系统及其制备方法,该多功能靶向给药系统具有良好生物相容性,生物可降解性,低毒性,能响应肿瘤细胞低ph环境、高谷胱甘肽浓度和cd44受体高度表达的生物特性释放药物,并且化疗与光热治疗、光动力治疗协同进行,从而明显提高对肿瘤细胞的杀伤效果,为肿瘤提供更好的治疗方法。
为了解决上述技术问题,本发明提供一种具有化疗光疗联合作用的多功能靶向给药系统的制备方法,包括以下步骤:
1)载药纳米粒的制备:先用溶剂挥发法将plga(乳酸-羟基乙酸共聚物)和ce6(二氢卟吩)溶解于丙酮中,避光将其滴加至去离子水中,避光搅拌除去丙酮,离心并用乙醇和去离子水依次洗涤除去游离的ce6,冷冻干燥得到ce6/plganp,将ce6/plganp分散于tris-hcl缓冲液(ph8.5,10mm)中,加入盐酸多巴胺,在避光条件下搅拌4~8h,然后将该混合液离心并用去离子水洗涤三次以除去未聚合的多巴胺,冷冻干燥得到ce6@pda;
2)ha-nh2的合成:用edc.hcl(1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐)和nhs(n-n-羟基琥珀酰亚胺)活化ha(透明质酸)的羧基,然后将其滴加到冰冷的含乙二胺的甲酰胺溶液中,搅拌反应24~48h,用过量冷丙酮中沉淀反应液并透析,得ha-nh2;
3)dox-ss-ha-nh2的合成:用edc和nhs活化3,3'-二硫代二丙酸的羧基,将盐酸阿霉素(dox)加到活化后的反应液中,避光搅拌24~48h得dox-ss-cooh,加入催化剂二乙胺,加入ha-nh2,避光搅拌反应24~48h,将所得反应液用过量丙酮沉淀,透析,冷冻干燥,得到dox-ss-ha-nh2;
4)靶向给药系统ce6@pda-ha-ss-dox的制备:称取步骤1)所得的ce6@pda与步骤3)所得的dox-ss-ha-nh2溶解于磷酸盐缓冲溶液(ph=9)中,室温下避光搅拌反应,离心洗涤,透析,冷冻干燥得ce6@pda-ha-ss-dox。
作为上述技术方案的优选,本发明提供的具有化疗光疗联合作用的多功能靶向给药系统的制备方法进一步包括下列技术特征的部分或全部:
作为上述技术方案的改进,所述步骤1)中ce6:plga的质量比=1:10~1:20,丙酮挥发搅拌时间为12~24h,多巴胺聚合反应温度在20~30℃,多巴胺聚合反应搅拌时间为4~8h。
作为上述技术方案的改进,所述步骤2)中反应温度在30~50℃;活化羧基时羧基:edc:nhs的摩尔比=1:2:2。
作为上述技术方案的改进,所述步骤3)中反应温度在40~60℃;二硫代二丙酸:盐酸阿霉素:ha-nh2(重复单元)的摩尔比=1:1:2~1:2:2。
作为上述技术方案的改进,所述步骤4)中ce6@pda与dox-ss-ha-nh2的质量比为2:1~4:1,反应时间为24~48h。
本发明所得的靶向给药系统是球状纳米粒结构,易通过肿瘤的高通透性和滞留效应将药物传递至肿瘤部位,其中所选用的天然高分子材料透明质酸ha,聚多巴胺pda,聚乳酸-羟基乙酸共聚物plga都具有良好的生物相容性,无毒性。
与现有技术相比,本发明的技术方案具有如下有益效果:
1、本发明所述的靶向给药系统,为解决单一化疗治疗效果差的缺点,利用化疗-光热治疗-光动力治疗的联合作用多重杀伤肿瘤组织及细胞,增强抗肿瘤疗效;
2、透明质酸可以特异性与肿瘤细胞上的cd44受体结合,使得药物主动靶向至肿瘤细胞内部,减少对正常细胞的毒副作用,可以解决多药耐药性问题;
3、ha与dox之间的二硫键和覆盖在ce6纳米球表面的pda使系统能响应肿瘤细胞内部低ph和高谷胱甘肽的环境快速释放药物。
上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,而可依照说明书的内容予以实施,并且为了让本发明的上述和其他目的、特征和优点能够更明显易懂,以下结合优选实施例,详细说明如下。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例的附图作简单地介绍。
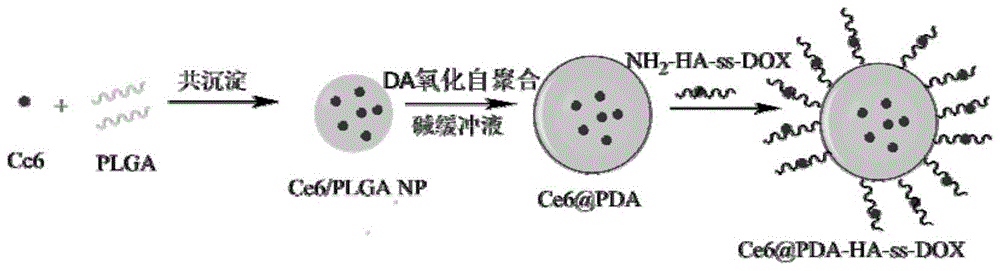
图1是本发明所述给药系统的制备示意图;
图2是本发明所述给药系统制备中的各物质的红外图谱;
图3为本发明所述ce6@pda和ce6@pda-ha的粒径分布图;
图4为本发明所述给药系统的药物释放曲线图。
具体实施方式
下面详细说明本发明的具体实施方式,其作为本说明书的一部分,通过实施例来说明本发明的原理,本发明的其他方面、特征及其优点通过该详细说明将会变得一目了然。
一种具有化疗光疗联合作用的多功能靶向给药系统的制备方法,
1)ce6@pda的制备
将20mgplga和2mgce6溶解于1ml丙酮中,避光将其滴加至不断搅拌的20ml去离子水中,搅拌12小时除去丙酮,离心并用去乙醇和离子水依次洗涤除去游离的ce6,冷冻干燥得到ce6/plganp,将50mgce6/plganp分散于50mltris-hcl缓冲液(ph8.5,10mm)中,加入50mg盐酸多巴胺,避光搅拌4小时,然后将该混合液离心并用去离子水洗涤三次以除去未聚合的多巴胺,冷冻干燥得到ce6@pda。
2)ha-nh2的合成
称取50mgha、145.6mgedc.hcl、88mgnhs,溶于15ml甲酰胺,氮气保护下活化ha的羧基,然后将其缓慢滴加到冰冷的含2.704mmol乙二胺的10ml甲酰胺溶液中,在氮气环境下搅拌反应24小时,用过量冷丙酮中沉淀反应液,透析72小时,得ha-nh2。
3)dox-ss-ha-nh2的制备
用99.7mgedc和59.8mgnhs活化27.34mg3,3'-二硫代二丙酸的羧基(羧基:edc:nhs(重复单元)的摩尔比=1:2:2),将37.68mg盐酸阿霉素加到活化后的反应液中,避光搅拌24小时得dox-ss-cooh,加入催化剂二乙胺,加入50mgha-nh2,避光搅拌反应24小时,将所得反应液用过量丙酮沉淀,透析,冷冻干燥,得到dox-ss-ha-nh2。
4)靶向给药系统ce6@pda-ha-ss-dox的制备
称取20mg步骤1)所得的ce6@pda与10mg步骤3)所得的dox-ss-ha-nh2溶解于10mlpbs缓冲溶液(ph=9)中,室温下避光搅拌反应24小时,离心洗涤,透析,冷冻干燥得ce6@pda-ha-ss-dox。
实施例2
1)ce6@pda的制备
将30mgplga和2mgce6溶解于1ml丙酮中,避光将其滴加至不断搅拌的20ml去离子水中,搅拌12小时除去丙酮,离心并用去乙醇和离子水依次洗涤除去游离的ce6,冷冻干燥得到ce6/plganp,将50mgce6/plganp分散于50mltris-hcl缓冲液(ph8.5,10mm)中,加入50mg盐酸多巴胺,避光搅拌6小时,然后将该混合液离心并用去离子水洗涤三次以除去未聚合的多巴胺,冷冻干燥得到ce6@pda。
2)ha-nh2的合成
称取50mgha、145.6mgedc.hcl、88mgnhs,溶于15ml甲酰胺,氮气保护下活化ha的羧基,然后将其缓慢滴加到冰冷的含2.704mmol乙二胺的10ml甲酰胺溶液中,在氮气环境下搅拌反应24小时,用过量冷丙酮中沉淀反应液,透析72小时,得ha-nh2。
3)dox-ss-ha-nh2的制备
用99.7mgedc和59.8mgnhs活化27.34mg3,3'-二硫代二丙酸的羧基,将37.68mg盐酸阿霉素加到活化后的反应液中,避光搅拌24小时得dox-ss-cooh,加入催化剂二乙胺,加入50mgha-nh2,避光搅拌反应36小时,将所得反应液用过量丙酮沉淀,透析3天,冷冻干燥,得到dox-ss-ha-nh2。
4)靶向给药系统ce6@pda-ha-ss-dox的制备
称取30mg步骤1)所得的ce6@pda与10mg步骤3)所得的dox-ss-ha-nh2溶解于10mlpbs缓冲溶液(ph=9)中,室温下避光搅拌反应24小时,离心洗涤,透析,冷冻干燥得ce6@pda-ha-ss-dox。
实施例3
1)ce6@pda的制备
将30mgplga和2mgce6溶解于1ml丙酮中,避光将其滴加至不断搅拌的20ml去离子水中,搅拌12小时除去丙酮,离心并用去乙醇和离子水依次洗涤除去游离的ce6,冷冻干燥得到ce6/plganp,将50mgce6/plganp分散于50mltris-hcl缓冲液(ph8.5,10mm)中,加入50mg盐酸多巴胺,避光搅拌8小时,然后将该混合液离心并用乙醇和去离子水依次洗涤以除去未聚合的多巴胺,冷冻干燥得到ce6@pda。
2)ha-nh2的合成
称取50mgha、145.6mgedc.hcl、88mgnhs,溶于15ml甲酰胺,氮气保护下活化ha的羧基,然后将其缓慢滴加到冰冷的含2.704mmol乙二胺的10ml甲酰胺溶液中,在氮气环境下搅拌反应24小时,用过量冷丙酮中沉淀反应液,透析72小时,得ha-nh2。
3)dox-ss-ha-nh2的制备
用99.7mgedc和59.8mgnhs活化27.34mg3,3'-二硫代二丙酸的羧基,将37.68mg盐酸阿霉素加到活化后的反应液中,避光搅拌24小时得dox-ss-cooh,加入催化剂二乙胺,加入50mgha-nh2,避光搅拌反应24小时,将所得反应液用过量丙酮沉淀,透析3天,冷冻干燥,得到dox-ss-ha-nh2。
4)靶向给药系统ce6@pda-ha-ss-dox的制备
称取40mg步骤1)所得的ce6@pda与10mg步骤3)所得的dox-ss-ha-nh2溶解于10ml磷酸盐缓冲溶液(ph=9)中,室温下避光搅拌反应24小时,离心洗涤,透析,冷冻干燥得ce6@pda-ha-ss-dox。
实施例4
1)ce6@pda的制备
将20mgplga和2mgce6溶解于1ml丙酮中,避光将其滴加至不断搅拌的20ml去离子水中,搅拌12小时除去丙酮,离心并用去乙醇和离子水依次洗涤除去游离的ce6,冷冻干燥得到ce6/plganp,将50mgce6/plganp分散于50mltris-hcl缓冲液(ph8.5,10mm)中,加入50mg盐酸多巴胺,避光搅拌6小时,然后将该混合液离心并用去离子水洗涤三次以除去未聚合的多巴胺,冷冻干燥得到ce6@pda。
2)ha-nh2的合成
称取50mgha、145.6mgedc.hcl、88mgnhs,溶于15ml甲酰胺,氮气保护下活化ha的羧基,然后将其缓慢滴加到冰冷的含2.704mmol乙二胺的10ml甲酰胺溶液中,在氮气环境下搅拌反应24小时,用过量冷丙酮中沉淀反应液,透析72小时,得ha-nh2。
3)dox-ss-ha-nh2的制备
用99.7mgedc和59.8mgnhs活化27.34mg3,3'-二硫代二丙酸的羧基,将37.68mg盐酸阿霉素加到活化后的反应液中,避光搅拌24小时得dox-ss-cooh,加入催化剂二乙胺,加入50mgha-nh2,避光搅拌反应24小时,将所得反应液用过量丙酮沉淀,透析,冷冻干燥,得到dox-ss-ha-nh2。
4)靶向给药系统ce6@pda-ha-ss-dox的制备
称取20mg步骤1)所得的ce6@pda与10mg步骤3)所得的dox-ss-ha-nh2溶解于10ml磷酸盐缓冲溶液(ph=9)中,室温下避光搅拌反应36小时,离心洗涤,透析,冷冻干燥得ce6@pda-ha-ss-dox。
附图1为给药系统的制备示意图。首先将ce6与plga通过纳米沉淀法制备ce6/plganp;再在ce6/plganp表面的氧化自聚合多巴胺来制备ce6@pda;然后nh2-ha-ss-dox上的-nh2与pda的羰基、共轭苯环发生席夫碱和迈克尔加成反应,从而将其共价连接到pda上得到ce6@pda-ha-ss-dox多功能靶向给药系统。
附图2为给药系统制备中的各物质的红外图谱。附图2(a)为ha-nh2,pda以及pda-ha的红外图谱:ha-nh2在1631.82cm-1处有脂肪族c=o的伸缩振动峰,证明有酰胺键存在;pda在1631.16cm-1处有苯环的峰,在1757cm-1处有芳香族c=o的峰,这说明多巴胺自聚合形成了醌;pda-ha在3421.15cm-1处有烷基的c-h峰,且峰较大,这能证明透明质酸的存在,而在1651.47cm-1处的峰证明有c=n结构,这说明聚多巴胺的c=o与ha-nh2的氨基反应生成了席夫碱(c=n),即pda与ha成功连接。附图2(b)为ha和ha-ss-dox的红外图谱:ha-ss-dox在1660.78cm-1和1556.92cm-1处的峰说明有苯环存在,证明dox成功连接到ha上,且1246.53cm-1处的酰胺ⅲ峰进一步证明dox与ha通过酰胺键连接。
附图3为ce6@pda和ce6@pda-ha的粒径分布图。如图所示,两者粒径分布较集中,只有一个单峰,说明形态均匀,ce6@pda的平均粒径为167.3nm,ce6@pda-ha的平均粒径为203nm。
附图4为药物释放曲线图。附图4(a)为dox的药物释放曲线图,附图(b)为ce6的药物释放曲线图。如图所示,两种药物在ph5.0+透明质酸酶+谷胱甘肽(ph5.0+hy+gsh)中释药最快,且释药率最高,这说明制备的给药系统能响应肿瘤的酸性、透明质酸酶和高谷胱甘肽浓度环境快速释放药物;而在ph7.4的中性环境(血液)中,系统释药缓慢且释药率最终只能到达20%左右,这说明系统能在血液环境中保持稳定,避免无效释药,减小对正常细胞的毒副作用。
本发明所列举的各原料,以及本发明各原料的上下限、区间取值,以及工艺参数(如温度、时间等)的上下限、区间取值都能实现本发明,在此不一一列举实施例。
以上所述是本发明的优选实施方式而已,当然不能以此来限定本发明之权利范围,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和变动,这些改进和变动也视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!