用于小麦外显子测序基因定位的探针设计方法及定位方法与流程
本发明涉及小麦功能基因定位技术领域,特别是涉及用于小麦外显子测序基因定位的探针设计方法及定位方法。
背景技术:
现有小麦全基因组测序bsa-seq,是对小麦基因组整体进行测序检测,由于小麦基因组十分庞大,成本太高。
现有小麦转录组测序bsr-seq使用的是基因表达的数据进行检测,受到样本组织、样本时期、环境等因素的影响,有非常大的数据偏好,基因表达数据本身不是遗传信息,具有一定的假阳性。
现有snp芯片无法获取基因的序列信息且snp密度相对低,snp芯片只能分析突变信号,无法获取到材料、样本的序列信息。
技术实现要素:
鉴于以上所述现有技术的缺点,本发明的目的在于提供一种用于小麦外显子测序基因定位的探针设计方法,用于解决现有技术中小麦基因组庞大导致使用全基因测序bsa-seq手段获得基因信息成本高昂的问题,同时,本发明还将提供一种小麦外显子测序基因定位方法。本发明中的一种用于小麦外显子测序基因定位的探针设计方法通过设计超高密度多重引物探针,在全基因组dna水平对小麦基因组超过16万个基因外显子进行特异性捕获,只针对于基因外显子序列进行测序,在同样的基因测序深度情况下,减少了80%以上的测序成本,小麦基因组15gb大小,全基因重测序30倍覆盖需要450g数据量,该方法仅需对300m区段进行测序,100倍覆盖需要数据量30g数据量。
为实现上述目的及其他相关目的,
本发明的第一方面,提供一种用于小麦外显子测序基因定位的探针设计方法,包括如下步骤:
步骤一、通过大规模转录组测序数据选取不同的组织、环境、发育等数据,再通过高通量测序平台进行测序,得到测序数据待用;
步骤二、通过star软件对测序数据与比对参考基因组进行比对分析,再通过stringtie函数进行转录本拼接,并过滤基因表达量tpm(转录本表达量transcriptsperkilobasemillion)小于2的转录本,保留基因表达量tpm≥2的转录本待用;
步骤三、将步骤二中的转录本与iwgsccsannotation转录本进行合并,得到合并后的转录本待用;
步骤四、通过transdecoder(v5.5.0)对步骤三中合并后的转录本进行orf(开放式阅读框openreadingframe)预测,再通过bedtools软件合并orf预测出的cds(编码区codingsequence)区域,再使用kmer算法扫描合并后的cds序列,分析重复及重叠的区域,去掉n及repeat区域的序列,得到筛选的序列;
步骤五、将步骤四中筛选的序列进行高密度探针合成,即得用于小麦外显子测序基因定位的探针。
上述探针设计方法通过设计超高密度多重引物探针,在全基因组dna水平对小麦基因组超过16万个基因外显子进行特异性捕获,只针对于基因外显子序列进行测序,在同样的基因测序深度情况下,减少了80%的测序成本。
步骤二中比对参考基因组具体为iwgscrefseq(https://www.wheatgenome.org/)。
于本发明的一实施例中,所述高通量测序平台为二代illumina或华大bgiseq。
于本发明的一实施例中,所述用于小麦外显子测序基因定位的探针为多重高密度磁珠探针。
本发明的第二方面,提供一种小麦外显子测序基因定位方法,包括如下步骤:
s1、将小麦dna打断成200-300bp长度的片段,再通过高通量测序文库(kapa、neb、illumina、诺唯赞等标准dna文库)构建dna测序预文库;
s2、再将打断后的小麦dna与上述探针进行杂交,再对磁珠进行富集从而获得与探针杂交的小麦dna序列,再使用洗脱液洗脱后得到小麦外显子dna序列待用;
s3、将s2步骤中小麦外显子dna序列进行高通量测序,对测序获得的数据使用标准分析流程进行变异检测,再通过统计方法进行定位即可。
全基因组测序需要对小麦16g基因组全部测序,需要的测序数据量是本发明中一种小麦外显子测序基因定位方法测序数据量的10倍以上,成本高昂。
上述小麦外显子测序基因定位方法无数据偏好,上述小麦外显子测序基因定位方法是在dna水平进行测序,避免了转录组bsr-seq方法只能对已经表达的基因进行分析的偏好。小麦外显子测序基因定位方法中设计捕获的目标区间超过260m,实际捕获区间超过300m,包含16万个基因的全部外显子及部分非编码启动子区域、调控区域,能够提供绝大多数性状变异的解析。
于本发明的一实施例中,所述小麦dna的制备过程为:将小麦自然杂交,突变体材料与亲本杂交,突变体材料与多样性亲本杂交后分别获得f2和f3分离群体,再将两个极端表型分别取10~50株,每个极端表型再取对应的根茎叶组织混合提取dna,再将两个极端表型的dna等量混合,即得小麦dna。
于本发明的一实施例中,每个所述极端表型在提取dna的过程中选取10~50株。
于本发明的一实施例中,所述统计方法为滑动窗口期望方差、滑动窗口t检验、滑动窗口fisher精确检验或snp-index。
如上所述,本发明的用于小麦外显子测序基因定位的探针设计方法及定位方法,具有以下有益效果:上述小麦外显子测序基因定位方法无数据偏好,上述小麦外显子测序基因定位方法是在dna水平进行测序,避免了转录组bsr-seq方法只能对已经表达的基因进行分析的偏好。小麦外显子测序基因定位方法中设计捕获的目标区间超过260m,实际捕获区间超过300m,包含16万个基因的全部外显子及部分非编码启动子区域、调控区域,能够提供绝大多数性状变异的解析。
附图说明
图1显示为本发明实施例1中外显子捕获实验流程图。
图2显示为本发明实施例1中捕获数据分析流程图。
图3显示为本发明实施例1中snp的密度分布图。
图4显示为现有技术bsr-seq获得snp的密度分布图。
图5显示为本发明实施例1中外显子测序基因定位结果图。
具体实施方式
以下由特定的具体实施例说明本发明的实施方式,熟悉此技术的人士可由本说明书所揭露的内容轻易地了解本发明的其他优点及功效。
实施例1
一种用于小麦外显子测序基因定位的探针设计方法,包括如下步骤:
步骤一、通过大规模转录组测序数据选取不同的组织、环境、发育的数据,再通过二代illumina高通量测序平台进行测序,得到测序数据待用;
步骤二、通过star软件对测序数据与比对参考基因组进行比对分析,再通过stringtie函数进行转录本拼接,并过滤基因表达量tpm小于2的转录本,保留tpm≥2的转录本待用;比对参考基因组具体为iwgscrefseq(https://www.wheatgenome.org/);
步骤三、将步骤二中的转录本与iwgsccsannotation转录本进行合并,得到合并后的转录本待用;
步骤四、通过transdecoder(v5.5.0)对步骤三中合并后的转录本进行orf预测,再通过bedtools软件合并orf预测出的cds区域,再使用kmer算法扫描合并后的cds序列,分析重复及重叠的区域,去掉n及repeat区域的序列,得到筛选的序列;
步骤五、将步骤四中筛选的序列进行高密度探针合成,即得用于小麦外显子测序基因定位的多重高密度磁珠探针。
一种小麦外显子测序基因定位方法,包括如下步骤:
s1、将小麦自然杂交,突变体材料与亲本杂交,突变体材料与多样性亲本杂交后分别获得f2和f3分离群体,再将两个极端表型分别取20株,每个极端表型再取对应的根茎叶组织混合提取dna,再将两个极端表型的dna等量混合,即得小麦dna;
s2、将s1步骤中的小麦dna随机打断成200-300bp长度的片段,再通过高通量测序文库(kapa标准dna文库)构建dna测序预文库;
s3、再将打断后的小麦dna与上述探针进行杂交,再对磁珠进行富集从而获得与探针杂交的小麦dna序列,再使用洗脱液洗脱后得到小麦外显子dna序列待用;
s4、将s3步骤中小麦外显子dna序列进行高通量测序,对测序获得的数据使用标准分析流程进行变异检测,再通过滑动窗口t检验统计进行定位即可。
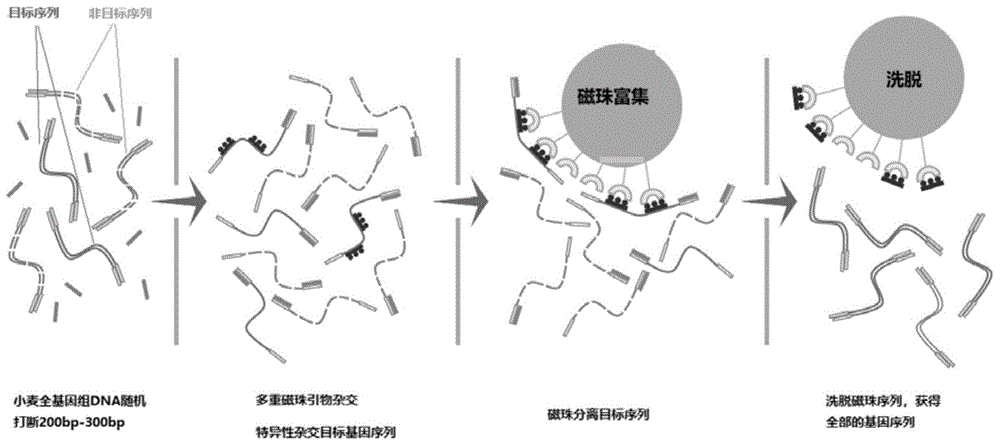
图1显示为外显子捕获实验流程,首先是小麦全基因组dna随机打断成200-300bp长度的片段,其中的目标序列与多重磁珠探针杂交变为特异性杂交目标基因序列,再通过磁珠富集使得特异性杂交目标基因序列被吸附,最后通过洗脱液洗脱后即可获得小麦外显子dna序列。
图2为捕获数据分析流程图,先进行小麦性状分离群体(回交分离群体、杂交分离群体、重组自交系分离群体等),再按照分离性状分别等量混合组织提取核酸或混合单株dna,再进行小麦外显子捕获测序,再进行bwa(burrows-wheeleraligner)序列对比,得到比对数据存储文件(bam格式),再采用gatk(genomeanalysistoolkit)软件进行基因组分析,得到gvcf或vcf格式的文件,再采用snpeff软件对gvcf或vcf格式的文件进行基因组注释,通过筛选(varbplot、candidateregion和varbscore)得到候选基因。
图3本实施例中获得snp的密度分布图,图4为现有bsr-seq获得snp密度分布图。从图中可以看出图3能够覆盖更多的基因组位置,且snp密度高出数倍。
图5为本发明实施例1中外显子测序基因定位结果图,从图中能够定位到非常小的候选区段,多数情况下直接定位到基因。该图片横轴为染色体位置,纵轴为每一个多态性区段的得分,图中的每一个点,代表一个多态性区段,点的得分越高,代表多态性区段越有可能为目标基因。
表格1为对捕获数据进行分析的结果:
表格1
实施例2
一种用于小麦外显子测序基因定位的探针设计方法,包括如下步骤:
步骤一、通过大规模转录组测序数据选取不同的组织、环境、发育的数据,再通过华大bigseq高通量测序平台进行测序,得到测序数据待用;
步骤二、通过star软件对测序数据与比对参考基因组进行比对分析,再通过stringtie函数进行转录本拼接,并过滤基因表达量tpm小于2的转录本,保留tpm≥2的转录本待用;比对参考基因组具体为iwgscrefseq(https://www.wheatgenome.org/);
步骤三、将步骤二中的转录本与iwgsccsannotation转录本进行合并,得到合并后的转录本待用;
步骤四、通过transdecoder(v5.5.0)对步骤三中合并后的转录本进行orf预测,再通过bedtools软件合并orf预测出的cds区域,再使用kmer算法扫描合并后的cds序列,分析重复及重叠的区域,去掉n及repeat区域的序列,得到筛选的序列;
步骤五、将步骤四中筛选的序列进行高密度探针合成,即得用于小麦外显子测序基因定位的多重高密度磁珠探针。
一种小麦外显子测序基因定位方法,包括如下步骤:
s1、将小麦自然杂交,突变体材料与亲本杂交,突变体材料与多样性亲本杂交后分别获得f2和f3分离群体,再将两个极端表型分别取30株,每个极端表型再取对应的根茎叶组织混合提取dna,再将两个极端表型的dna等量混合,即得小麦dna;
s2、将s1步骤中的小麦dna随机打断成200-300bp长度的片段,再通过高通量测序文库(illumina标准dna文库)构建dna测序预文库;
s3、再将打断后的小麦dna与上述探针进行杂交,再对磁珠进行富集从而获得与探针杂交的小麦dna序列,再使用洗脱液洗脱后得到小麦外显子dna序列待用;
s4、将s3步骤中小麦外显子dna序列进行高通量测序,对测序获得的数据使用标准分析流程进行变异检测,再通过滑动窗口fisher精确检验统计进行定位即可。
综上所述,本发明通过设计超高密度多重引物探针,在全基因组dna水平对小麦基因组超过16万个基因外显子进行特异性捕获,只针对于基因外显子序列进行测序,在同样的基因测序深度情况下,减少了80%的测序成本,小麦基因组15gb大小,全基因重测序30倍覆盖需要450g数据量,该方法仅需对300m区段进行测序,100倍覆盖需要数据量30g数据量。所以,本发明有效克服了现有技术中的种种缺点而具高度产业利用价值。
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种用于评估皮肤抗过敏情况的引物和探针及试剂盒的制作方法
- 一种用于评估皮肤防御情况的引物和探针及试剂盒的制作方法
- 一种荧光定量PCR检测猪delta冠状病毒的引物、探针、试剂盒及方法
- 一种快速区分HP-PRRS疫苗GDr180株与野毒株的引物、探针及方法
- 一种用于检测样品中猪凸隆病毒的荧光rt-pcr引物和探针和试剂盒及检测方法
- 一种阿胶中驴、马、牛和猪源性巢式荧光pcr检测引物、探针、试剂盒、检测方法及应用
- 一种用于评估皮肤防晒情况的引物和探针及试剂盒的制作方法
- 一种用于评估皮肤抗皱情况的引物和探针及试剂盒的制作方法
- 一种用于检测car-t转导效率的引物、探针和方法
- 用于微嵌合检测和个体识别的引物、探针、试剂盒及方法