一种基于正则化变分自动编码器的药物分子生成方法与流程
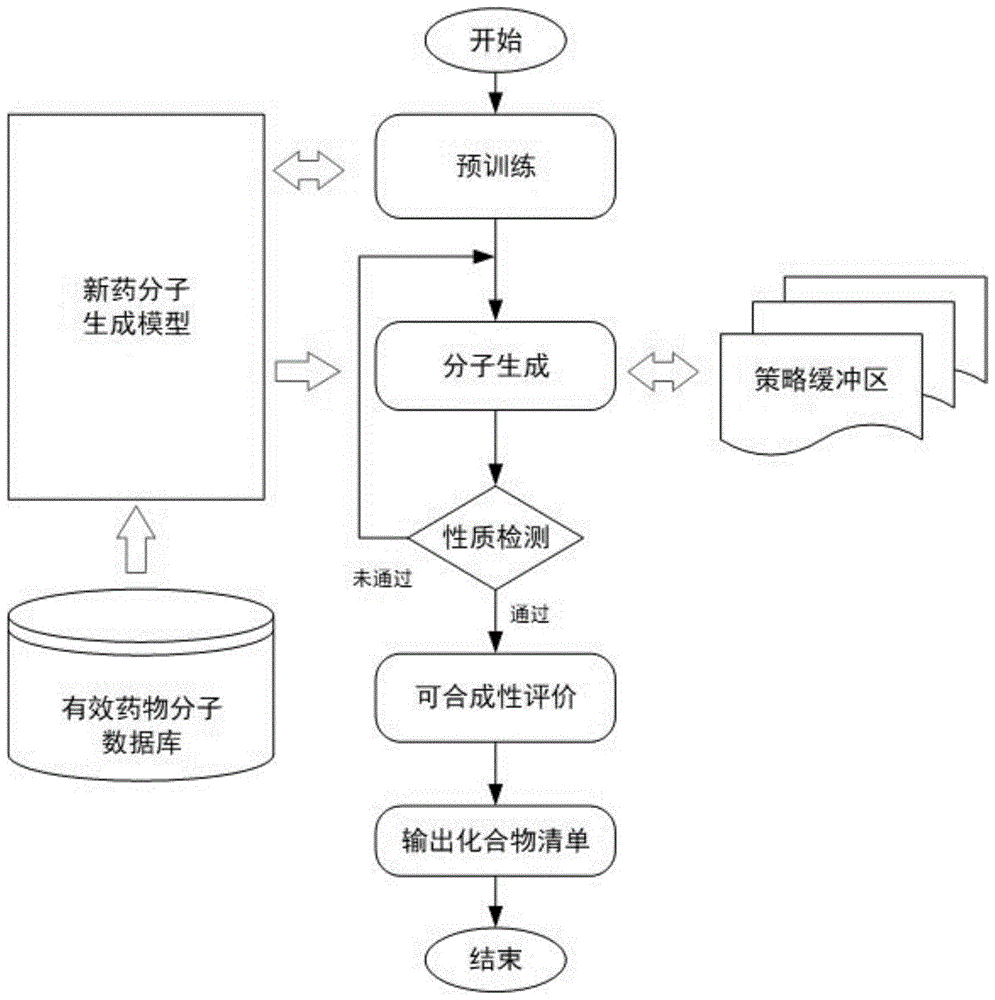
本发明涉及计算机人工智能与新药分子设计的交叉技术领域,尤其涉及一种基于正则化变分自动编码器的药物分子生成方法,是一种基于图神经网络、深度生成模型和性质目标正则化的进行新药分子设计的方法,适用于新药发现过程中候选药物分子的设计与生成。
背景技术:
新药研发耗资巨大、周期漫长且成功率很低。其中候选药物分子的筛选是前期的关键环节,计算机辅助设计以及最新人工智能技术的引入,已经大幅度提升了分子筛选的效率。但传统的计算机筛选方法大都针对已有的化合物,或基于结构或基于性质等特征对其进行筛选,新一代的新药发现策略则开启了直接设计全新分子的途径。具体是指,针对某种疾病或靶点预设药效等对新分子的期望属性,根据已有相关药物分子的结构,采用计算机模拟生成的计算方法设计全新的分子结构,所依据的原理是“相似的结构很可能具有相同的性质”,进而通过化学合成方法以人工方式合成新分子,并进一步在真实的化学、生物和人体等环境中检测其药效和其他物理化学性质,从而完成新药的开发。该途径可大幅缩短新药研制和临床试验的时间,具有广阔的应用前景。
在上述过程中,通过计算机模拟、生成具备特定生化性质的药物分子是此类方法的关键。然而,现有药物分子生成方法仍面临一些挑战。首先,潜在药物分子的表示空间巨大,而且不连续,搜索药物分子任务本身十分艰巨。有医疗文献显示,化学分子的表示空间范围可达1023~1060。其次,分子结构和分子性质之间的对应关系十分微妙,难以进行准确的量化描述,即使结构十分接近的分子其生化性质也可能完全不同,即,对某些子结构的微小改动都可以导致性质的大幅改变。
现有分子生成方法一般采用smiles(simplifiedmolecularinputlineentryspecification,简化分子线性输入规范)和分子指纹等字符串方式表示分子,并借助自然语言处理的方法来实现生成算法,此类方法的鲁棒性较差,即微小的修改可能导致完全不合理的分子。同时,基于字符串的语法约束会对分子生成产生过多限制,给分子生成的优化过程造成负担。在生成分子图的任务中,一个关键的难点在于使得模型的学习目标包含高度复杂而不可微的分子性质度量,为了解决这个问题,目前有基于强化学习和基于贝叶斯优化等两种方法。但是,基于强化学习的方法需要引入额外网络,使得计算和收敛难度增大,同时很难设计合理的即时奖励;基于贝叶斯优化的方法由于其两阶段的特性,使得性质目标的优化很大程度依赖于第一阶段中所学模型的隐向量空间的光滑性。
技术实现要素:
本发明提出了一种基于正则化变分自动编码器的药物分子生成方法。该方法采用图(graph)数据结构(图矩阵)表示药物分子,以变分自动编码器作为基本生成框架,在此基础上加入性质目标正则化项。利用图数据结构表示药物分子,在结构上,该模型包括编码器和解码器两部分,其中编码器利用图神经网络直接对输入图表示进行编码,解码器主要由多层感知机(多层全连接神经网络)组成,优化目标包括重建损失,kl损失(kullback-leiblerdivergence,kl散度),以及性质正则化损失三部分。其中,性质正则化损失采用蒙特卡洛采样进行估计。采用本发明方法可有效生成分子性质优化的候选药物分子。
本发明方法直接形式化性质目标正则化项来促使模型学习高度复杂而不可微的性质度量,有效降低了网络复杂度且无两阶段操作,对隐向量空间光滑性的依赖性降低,分子生成效果较优。
本发明的技术方案是:
一种基于正则化变分自动编码器的药物分子生成方法,利用变分自动编码器,将药物分子表示为图矩阵数据,建立包括编码器和解码器的药物分子生成模型,其中编码器利用图神经网络直接对输入的药物分子图进行编码,解码器包括多层感知机,优化目标包括重建损失、kl(kullback-leiblerdivergence,kl散度)损失及性质正则化损失,其中性质正则化损失利用蒙特卡洛采样进行估计;包括以下主要步骤:构建有效药物分子库、搭建药物分子生成的基本模型、设计并实现基于图数据结构的深度生成模型、设计并实现性质目标正则化方案、模型训练、生成流程执行、生成结果的验证与应用。
步骤1,构建有效药物分子库
通过有针对性地收集现有药物的信息,即真实数据,建立用于药物分子生成模型训练和测试的分子信息库,分子信息库包含了一些已知药物分子的结构信息(药物分子表示为图数据,其结构信息包括图中的节点和边),和用于测试的药物分子的理化信息,例如分子的脂水分配系数的对数值(logp)、类药性、可合成性等。
步骤2,搭建药物分子生成模型
21)搭建药物分子生成过程的基本模型,即变分自动编码器,通过学习真实数据和先验分布的联合概率分布实现分子生成。
本发明构建药物分子生成模型pθ(g|z),采用图数据(g)表示药物分子(z为g通过编码器编码得到的隐向量),在变分推断中,利用变分后验qφ(z|g)来估计真实后验,最大化药物分子生成模型pθ(g|z)如下目标函数:
式(1)中,
22)设计并实现基于图数据结构的深度生成模型
设计基于图神经网络的编码器和图数据结构解码器,使得模型以图数据结构为表示进行计算。药物分子生成模型包括编码器和解码器,可分别采用图神经网络和全连接神经网络。具体地:
在式(1)中,图数据g用于表示真实分子,模型训练数据采用步骤1建立的药物分子库中的药物分子;z为g通过编码器编码得到的隐向量。编码器输出一个矩阵
23)设计并实现药物分子性质目标正则化方法
设s是需要优化的药物分子性质,将药物分子性质关于分布pθ(g|z)的期望作为正则化项。该期望表示为式(2):
其中,sg表示图数据g对应的药物分子的待优化的性质s,表示为式(3):
其中,o是所有子图模式的集合,no是模式o在图g中出现的次数,co是模式o每次出现对于该性质的贡献。结合公式(2)和(3)可得式(4):
式(4)中,
基于上述药物分子性质目标正则化方法,所提出的药物分子生成模型能够生成具有特定性质的分子。
步骤3,药物分子生成模型的训练
为训练所设计的药物分子生成模型,需要依据合理的目标函数。本发明中,药物分子生成模型需最大化如下目标函数
其中,
其中,
通过上述训练过程利用步骤1建立的药物分子库中的药物分子对药物分子生成模型pθ(g|z)进行训练,得到训练好的药物分子生成模型pθ(g|z)。
步骤4,利用训练好的药物分子生成模型pθ(g|z),执行药物分子生成流程,生成候选药物分子
设定药物分子的待优化目标,利用上述从先验分布pθ(z)中采样再经过训练后得到的训练好的药物分子生成模型pθ(g|z),从先验分布中采样并送入生成模型生成候选药物分子,输入候选药物分子,将候选药物分子与药物分子库中具有理化信息的分子进行比较,用于测试生成的药物分子。
步骤5,性质检测
对生成的候选药物分子可进行药物分子性质检测(包括药物分子的生物利用度)和可合成性评估,进一步确认其有效性。
通过上述步骤,实现一种基于正则化变分自动编码器的药物分子生成。
与现有技术相比,本发明的有益效果是:
分子生成的目标之一是优化分子的性质,本发明采用的是基于图表示的分子生成方法,图中的节点用于表示原子,图中的边用于表示化学键,该方法比基于字符串的方法更具可解释性和鲁棒性。本发明形式化了一个反应定量分子性质的正则化项,利用蒙特卡洛方法可以得到一个可微的估计值,使得可以用梯度下降法来学习此目标,省去了额外的网络设计,降低了收敛难度,同时生成阶段可直接解码先验分布,从而简化了基于贝叶斯优化类方法的两步操作。采用本发明技术生成的候选药物分子有效性更高,分子的性质更优异。
附图说明
图1为本发明提供的药物分子生成方法的流程框图。
图2为本发明实施例的药物分子生成模型示意图。
图3为本发明的训练示意图
图4为本发明的生成示意图
具体实施方式
下面结合附图,通过实施例进一步描述本发明,但不以任何方式限制本发明的范围。
新药研发耗资大、周期长,其关键环节之一是候选药物分子的筛选,人工智能技术的引入可以有效提升筛选效率,但基于筛选的方法限于现有已有化合物,范围有限,新一代方法则侧重全新的分子生成方法。
本发明提出基于深度生成模型的分子生成模型,涉及计算机人工智能和医药分子设计的交叉领域,其核心思想是将图神经网络和性质正则化同时引入深度生成模型,能够有效利用图表示的符合直觉同时能够捕捉分子内在相似性等特点,并解决了分子性质作为优化目标高度复杂而不可微的问题。
如图1所示,本发明方法包括构建有效药物分子库、搭建药物分子生成的基本模型、设计并实现多任务强化学习模块、设计并实现对抗模仿学习模块、模型预训练、生成流程执行、生成结果的验证与应用等步骤。
图2所示为本发明具体实施时构建的基于深度生成模型的药物分子生成模型的结构示意图,其中,图2的上半部分为训练阶段的数据流通路径,真实药物分子a(如阿司匹林)
经过编码器编码后得到变分后验概率分布qφ(z|g)=n([-0.1159,-0.2748,-0.0782,-0.0107,0.0685,0.0596,-0.1988,0.0028],[0.0154,0.5714,0.9075,0.9758,0.9763,1.0359,0.7208,0.9160]×i),此分布再经过解码之后得到解码分子b
在训练阶段我们的目标是使得解码得到的分子b和输入的分子a相同,同时迫使qφ(z|g)与pθ(z)具有相同的分布,详见图3。图2的下半部分为生成阶段的数据流通路径,在解码器训练完成之后,为了生成有效的药物分子,我们从先验分布中采样得到分子的隐向量表示,然后将其送入解码器即可生成我们想要的分子c(即候选药物分子)
详见图4。
步骤1构建有效药物分子库
收集现有药物分子的信息,包括结构、物理化学属性、药效等,建立用于药物分子生成的分子信息库,并标注各种药物对于特定病种的有效性。药物分子库将用于模型的训练和测试。模型训练时可利用药物分子库中的分子图数据,模型测试时将待测药物分子与药物分子库中具有理化信息的分子进行比较。
步骤2搭建药物分子生成基本模型
本方法是基于深度生成模型的一种分子图生成方法,我们的目标是学习一个药物分子生成模型pθ(g|z)。在变分推断中,我们利用一个变分后验qφ(z|g)来估计真实后验,根据变分推断的原理,最大化如下目标函数:
式(1)中,
步骤3设计并实现基于图数据结构的深度生成模型,作为药物分子生成模型;
药物分子生成模型包括编码器和解码器,可分别采用图神经网络和全连接神经网络。具体地:
在公式(1)中,图数据g用于表示真实分子,z为g通过编码器编码得到的隐向量。编码器输出一个矩阵
步骤4设计并实现分子性质目标正则化方法,使得模型能够生成具有特定性质的分子;
设s是需要优化的分子性质(如分子的脂水分配系数的对数值(logp)、类药性、可合成性等性质),我们可以将它关于分布pθ(g|z)的期望作为正则化项。该期望可以写成式(2):
其中,sg是表示为图数据g的药物分子待优化的性质s。同时,根据结构性质关系模型,得到式(3):
其中,q是所有模式的集合,nq是模式q在图g中出现的次数,cq是模式q每次出现对于该性质的贡献。结合公式(2)和(3)可得式(4):
式(4)中,
步骤5模型训练
本发明中的药物分子生成模型最大化如下目标函数
我们采用蒙特卡洛估计来计算
其中,v(m)和e(m)分别是采样所得模式q(m)中的原子集合和边集合,pit和pijr分别表示it和ijr对应的神经元值。
步骤6生成流程执行
根据给定的优化目标,从先验分布pθ(z)中采样,利用经过训练后得到的训练好的药物分子生成模型pθ(g|z),即可生成具有特定性质的药物分子。
步骤7生成结果的验证与应用
1)性质检测
对药物分子生成模型所生成的候选分子,进行性质检测。即根据分子结构计算并预测相应的分子性质,并将所得性质与设计要求做对比。具体地,一般为了得到有效的药物分子,对生成的药物分子的以下性质进行检测,判断药物分子的生物利用度。如选用lipinski规则检测:分子的分子量是否小于500,氢键给体(包括羟基、氨基等)的数量不超过5个,氢键受体的数量不超过10个,脂水分配系数的对数值(logp)在-2到5之间,其可旋转键的数量不超过10个等。对于满足要求的分子,将进一步拆分其结构得到状态序列,并将其加入到策略缓冲区中,同时将其移交至下一步做可合成性检验。对于尚未达到要求或可能有悖于药物分子规律的分子,将其转入下一轮的生成过程,进行再次迭代,或放弃后开启新一轮的生成过程。
2)可合成性评估
鉴于本模型所生成的分子很可能并不存在于现实世界,因此,为评估后续化学工作者合成该分子的难易程度,需要对其可合成性进行评估。本发明采用逆合成分析法进行分子的可合成性评估。即采用互换、添加官能团以及逆向切断等方法将生成的分子变成若干中间产物或原料。之后再对中间产物重复进行逆合成分析,直到所有中间产物变成简单、易得的分子。
3)为实体试验输出候选结果
输出本方案所生成的分子,即作为新药的候选分子输出其结构描述。本发明技术方案可用于辅助新药研发,能够提高其对候选分子的筛选效率,但是并不能取代新药研发各项实体试验,包括生化试验、动物试验和各期临床试验等,新药研发机构需要完成后续试验,以得到最终的新药化合物。
计算机辅助分子生成是一个新兴的交叉领域,在医药研发、疾病诊断和材料科学中具有广阔的应用前景。一方面,传统药物分子的研发周期漫长、耗资巨大且很难筛选出有效的分子。虽然计算机辅助设计及人工智能算法极大的提高了分子筛选的效率。但传统的计算机大多依赖已有的化合物、结构、性质进行设计,而新一代的药物设计往往是针对疾病或靶点预设药效等对药分子的期望属性,根据相关已有的药分子结构进行新药分子的设计。而本发明所提出的对抗模仿学习方法可以很好的学习到分子结构与特定药物属性之间难以量化的关系,能以更高的效率生成可满足新药设计需求的候选药物分子,且其鲁棒性、生成分子的新颖性均可获得提升。
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
- 还没有人留言评论。精彩留言会获得点赞!