蛋白质复合物深度多序列比对方法与流程
本发明涉及生物信息学中蛋白质复合物深度多序列比对领域,具体地说是一种认知蛋白质单体序列与其构成复合物序列蛋白质之间蛋白质家族相似性关系的方法。
背景技术:
生物信息学是一门生物学与信息科学交叉而形成的年轻学科,它是当今生命科学和自然科学的重大前沿领域之一,其研究重点主要体现在基因组学和蛋白质组学两方面。生物信息学的研究对于加深对人类生命过程的认知,帮助人们改善其生存环境和提高生活质量有着重要意义,得到了国内外学者广泛重视。
蛋白质作为生命现象的物质基础之一,是构成一切细胞组织结构的重要组成成分,参与了生物体内许多方面的重要生命过程,是生命活动的重要承担者。虽然说脱氧核糖核酸(dna)是遗传信息的载体,但是遗传信息的复制、转录和表达都需要依靠各种蛋白质之间的协作才能完成。蛋白质组学较之于基因组学,对于生命现象的解释更直接、更准确,近年得到了快速发展,并受到世界各国学者的高度关注。在后基因组时代,随着蛋白质测序技术的飞速发展,蛋白质序列的数据呈爆炸性增长,目前在著名的蛋白质数据库uniprotkb中,已经存有超过120,243,849条蛋白质的一级序列信息(截止到2018-07-16),并继续保持快速增长的趋势。然而,面对如此海量的蛋白质序列信息,以当前已测序的蛋白质数据为例,0.1%(140,000)的蛋白质被求解出三维结构,0.3%的真实蛋白质复合物被实验验证并解出三维结构,收录在著名的蛋白质结构数据库pdb中。这个差距更会随着技术的不断进步和成熟而不断扩大。
通过文献阅读,可以发现在蛋白质单体序列多序列比对领域已经取得了丰硕的成果,发表了不少具有很高理论意义以及实用价值的论文,经典的蛋白质单体序列比对方法有blast(kent,w.james."blat—theblast-likealignmenttool."genomeresearch12.4(2002):656-664.)、psi-blast(altschul,stephenf.,etal."gappedblastandpsi-blast:anewgenerationofproteindatabasesearchprograms."nucleicacidsresearch25.17(1997):3389-3402.)、hhblits(remmert,michael,etal."hhblits:lightning-fastiterativeproteinsequencesearchingbyhmm-hmmalignment."naturemethods9.2(2012):173.)以及jackhhmer(kandulski,a.,etal."jackhammeresophagus:high-resolutionmanometryandtherapeuticapproachusingperoralendoscopicmyotomy(poem)."diseasesoftheesophagus29.6(2016):695-696.)。但是,通过进一步的梳理分析,当前的研究工作还主要研究如何提升蛋白质单体多序列比对质量,对于蛋白质复合物多序列比对的仅仅简单的沿用蛋白质单体多序列比对方法,例如complexcontact(zeng,hong,etal."complexcontact:awebserverforinter-proteincontactpredictionusingdeeplearning."nucleicacidsresearch46.w1(2018):w432-w437.)、gremlins(seoane,joan."gremlinssabotagethemechanismsofcancerstemcelldifferentiation."cancercell25.6(2014):716-717.)、evfold(hopf,thomasa.,etal."sequenceco-evolutiongives3dcontactsandstructuresofproteincomplexes."elife3(2014):e03430.)。
尽管这些研究虽然能够用来用了作为蛋白质复合物多序列比对方法,但挑战仍然存在。首先,这些方法的重点都蛋白质复合物的二级结构预测,因此其多序列比对结果精度不高。其次,使用单一的策略进行构建蛋白质复合物多序列比对数据库,容易造成多序列比对结果有且仅有一条查询序列,从而导致多序列比对精度差。此外,机械性的将不同数据库进行结合用来构建蛋白质复合物数据库,导致不同数据库之间的比对结果相互影响,从而限制了多序列比对的精度提升。
技术实现要素:
本发明的目的在于提供一种具有多序列比对结果质量高、深度大、序列来源范围广以及泛化能力强的蛋白质复合物多序列比对方法,用于解决上述蛋白质复合物多序列比对中由于数据库单一以及搜索深度低而导蛋白质复合物多序列比对结果质量偏低的缺点。
实现本发明目的的技术解决方案为:一种蛋白质复合物多序列比对方法,包括以下步骤:
步骤1、蛋白质单体序列数据库与基因组距离搜索算法的构建:首先,从蛋白质单体序列全基因组数据中下载uniclust30蛋白质单体序列数据库。其次,使用多序列比对软件hhblits软件分别对蛋白质单体序列搜索蛋白质序列数据库uniclust30进行多序列比对,得到蛋白质单体序列的多序列比对信息。再次,分别将蛋白质单体序列的多序列比对结果与基因组数据库(ena)进行比对。最后,根据两个不同的单体序列的多序列比对与基因组数据库种的比对的距离将不同的两种不同单体序列的多序列比对进行连接,从而得到蛋白质复合物基于基因组距离的多序列比对;
步骤2、蛋白质单体序列数据库与物种相似性搜索算法的构建:首先,从公共数据库美国国立信息中心(ncbi)中下载物种分类数据库(taxonomy)。其次,使用多序列比对软件hhblits软件分别对蛋白质单体序列搜索步骤1构建的蛋白质序列数据库uniclust30进行多序列比对,得到蛋白质单体序列的多序列比对信息。其次,分别将蛋白质单体序列的多序列比对结果与物种分类数据库(taxonomy)进行物种比对。最后,根据物种比对结果将两个不同的单体多序列比对进行连接,从而得到蛋白质复合物基于物种的多序列比对结果;
步骤3、蛋白质交互网络数据库与蛋白质相互作用搜索算法构建:首先,从公共数据库蛋白质相互作用网络数据库(string)中下载蛋白质相互作用信息(stringlinker)以及蛋白质相互作用的序列信息数据库(stringdatabase)。其次,使用多序列比对软件hhblits软件分别对蛋白质单体序列搜索上述步骤构建的蛋白质相互作用序列信息数据库进行多序列比对,得到蛋白质单体序列的多序列比对信息。最后,根据蛋白质相互作用信息将两种不同蛋白质单体序列的多序列比对进行连接,从而得到基于蛋白质相互作用网络的蛋白质复合物序列的多序列比对结果;
步骤4、蛋白质复合物多序列比对方法选择:首先,使用计算步骤1中基于基因组距离的蛋白质复合物多序列比对中有效序列的条数。其次,如果步骤1中的多序列比对中的序列条数满足要求,则将步骤1的对序列比对作为去除冗余序列步骤的输入。反之则将步骤1中的多序列比对与步骤2中基于物种类别的多序列比对进行合并并且计算有效序列的条数。再次,如果步骤1与步骤2的合并后的有效序列条数满足条件,则将合并结果作为除冗余序列步骤的输入。否则,将步骤1与步骤2以及步骤3中基于蛋白质相互作用网络的多序列比对合并,作为去除冗余序列步骤的输入;
步骤5、去除多序列比对冗余:将步骤4中产生的蛋白质复合物多序列比对进行去除冗余,使得去除冗余后的多序列比对中任意两条序列的相似性小于90%。
步骤6、在线预测:给定一个预测的蛋白质复合物序列,利用步骤1-5中的方法生成相应的蛋白质复合物序列的多序列比对,并将此蛋白质复合物的多序列比对结果以页面或者邮件的形似返回给用户,方便研究者使用。
本发明与现有技术相比,其显著优点:(1)提高了多序列比对深度:首先,多序列比对的深度是指层次深,而不仅仅是使用单一的搜索算法或者数据库进行比对;其次,不同层次的多序列比对方法不是机械的组合在一起,而是根据上一层的多序列比对结果中的有效序列个数进行判断,从而优化了多序列比对速度;最后,对不同层次的多序列比对结果进行去除冗余序列,以保证各层次融合后的序列具有特异性。(2)提高了多序列比对的质量:使用了不同的蛋白质单体数据库以及采用三种不同的策略对蛋白质复合物中的单体多序列比对结果进行连接,从而避免了使用一种单一的策略造成两个蛋白质单体序列无法进行连接,从而导致构建蛋白质复合物多序列比对失败。因此,采用三种不同的搜索连接策略,保证了对序列比对的结果,扩大的多序列比对的数据库范围,从而提升了多序列比对的质量。(3)提升了模型的泛化能力:使用了三种不同蛋白质单体多序列比对连接策略(基于基因距离,物种种类以及蛋白质交互网络),因此保证了对于任意一查询蛋白质复合物序列,都能产生其多序列比对结果。故本发明提升了模型的泛化能力。
附图说明
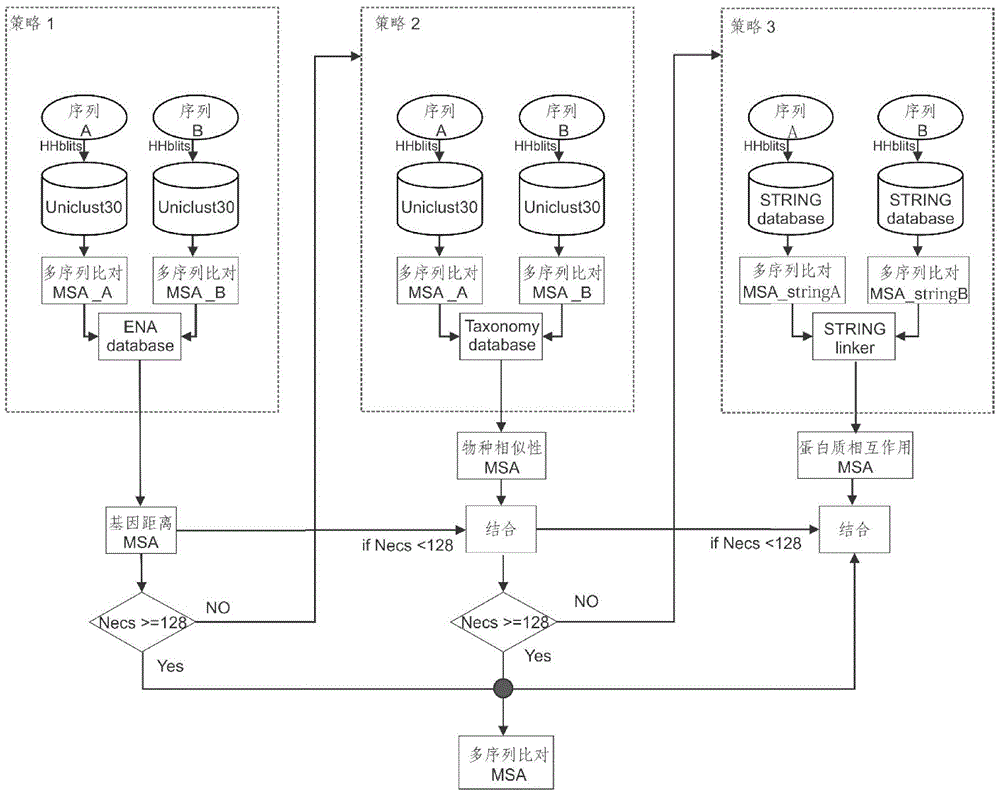
附图1为本发明的蛋白质复合物深度多序列比对方法的示意图。
具体实施方式
下面结合附图对本发明作进一步说明。
附图给出了本发明的多序列比对方法系统结构示意图。结合附图所示,根据本发明的实施例,一种蛋白质复合物深度多序列比对方法,包括了以下步骤:首先,构建蛋白质单体序列数据库。并且使用多序列比对软件搜索数据库得到多序列比对结果,然后根据基因距离信息将蛋白质单体连接;其次,将蛋白质单体序列的多序列比对结果与物种分类数据库(taxonomy)进行物种比对。根据物种比对结果将两个不同的单体多序列比对进行连接;再次,构建蛋白质相互作用信息(stringlinker)以及蛋白质相互作用的序列信息数据库(stringdatabase)。并且使用多序列比对软件hhblits软件分别对蛋白质单体序列搜索上述步骤构建的蛋白质相互作用序列信息数据库进行多序列比对,得到蛋白质单体序列的多序列比对信息。根据蛋白质相互作用信息将两种不同蛋白质单体序列的多序列比对进行连接;然后,根据不同策略的有效序列条数进行判断是否进行下一层次策略的多序列比对构建;由此,将上述步骤中产生的蛋白质复合物多序列比对进行去除冗余,使得去除冗余后的多序列比对中任意两条序列的相似性小于90%。最后,给定一个预测的蛋白质复合物序列,利用步骤1-5中的方法生成相应的蛋白质复合物序列的多序列比对,并将此蛋白质复合物的多序列比对结果以页面或者邮件的形似返回给用户,方便研究者使用。;下面将结合附图所示,更加具体地描述前述过程。
步骤1、蛋白质单体序列数据库与基因组距离搜索算法的构建:
给定一个蛋白质复合物数,其包含两条单体序列,序列a和序列b。然后分别对其序列a和序列b的使用多序列比对搜索算法搜索蛋白质数据库,然后根据基因距离信息进行构建策略1的多序列比对,具体步骤如下:
(1)从蛋白质单体序列全基因组数据(https://uniclust.mmseqs.com/)中下载uniclust30蛋白质单体序列数据库;
(2)分别序列a和序列b使用多序列比对软件hhblits软件搜索蛋白质序列数据库uniclust30进行多序列比对,分别得序列a和序列b的多序列比对信息msa_a和msa_b;
(3)将多序列比对结果msa_a和msa_b与基因组数据库进行比对,分别多序列比对结果的基因信息msa_a_gene和msa_b_gene;
(4)计算msa_a_gene与msa_b_gene中具有相同基因的两个蛋白质i和j的基因距离δgene,如果1≤δgene≤20,则将蛋白质i与蛋白质j连接起来;
(5)根据(1)~(4)的步骤,构建基于基因距离的蛋白质复合物多序列比对(msa)。
步骤2、单体序列数据库与物种相似性搜索算法的构建
(1)从公共数据库美国国立信息中心(ncbi)中下载物种分类数据库(taxonomy)
(2)将步骤1中序列a和序列b的多序列比对信息msa_a和msa_b分别与物种分类数据库(taxonomy)进行物种比对,并且分别得到msa_a和msa_b中蛋白质的物种信息;
(3)分别对msa_a和msa_b中的每个物种中蛋白质与查询序列的相似性从高到低进行排序;
(4)令p1,p2,…,pm为在msa_a中的特定物种根据序列相似性排序后的蛋白质,并且,q1,q2,…,qn为在msa_b中的特定物种根据序列相似性排序后的蛋白质。然后将pi与qi进行连接,其中,i=min(m,n)。
步骤3、蛋白质交互网络数据库与蛋白质相互作用搜索算法构建
(1)从公共数据库蛋白质相互作用网络数据库(string)中下载蛋白质相互作用信息(stringlinker)以及蛋白质相互作用的序列信息数据库(stringdatabase);
(2)使用多序列比对软件hhblits软件分别对序列a和序列b搜索蛋白质相互作用序列信息数据库进行多序列比对,分别得到多序列比对信息msa_stringa和msa_stringb。
(3)最后,根据蛋白质相互作用信息判断msa_stringa与msa_stringb中的任意两个蛋白质是否具有相互作用。如果有相互作用,则将二者连接起来。
步骤4、蛋白质复合物多序列比对方法选择
计算多序列比对中有效序列的个数
其中,l是蛋白质复合的链长,n是在蛋白质复合物多序列比对(msa)中序列的条数,sia,ja是序列i中的链a与序列j中链a的序列相似性得分,sib,jb是序列i中的链b与序列j中链b的序列相似性得分。另外,优化后的necs值为128。即,如果necs≥176则不进行下一策略的比对,否则继续。
使用蛋白质结构聚类算法软件spicker对上述步骤中产生的副本进行聚类,计算每一类中所有构象中原子坐标的平均值。所得到的原子坐标平均值作为聚类中心构象的原子坐标。
步骤5、去除多序列比对冗余
将步骤4中产生的蛋白质复合物多序列比对进行去除冗余,使得去除冗余后的多序列比对中任意两条序列的相似性小于90%。
步骤6、在线预测
对给定的预测蛋白质复合物序列,利用步骤1-5中的方法生成相应的蛋白质复合物三维结构,并将此蛋白质三维结构以页面或者邮件的形似返回给用户,方便研究者使用。
综上所述,首先,多序列比对的深度是指层次深,而不仅仅是使用单一的搜索算法或者数据库进行比对;其次,不同层次的多序列比对方法不是机械的组合在一起,而是根据上一层的多序列比对结果中的有效序列个数进行判断,从而优化了多序列比对速度;最后,对不同层次的多序列比对结果进行去除冗余序列,以保证各层次融合后的序列具有特异性。故本发明提升了多序列比对的深度;其次,使用了不同的蛋白质单体数据库以及采用三种不同的策略对蛋白质复合物中的单体多序列比对结果进行连接,从而避免了使用一种单一的策略造成两个蛋白质单体序列无法进行连接,从而导致构建蛋白质复合物多序列比对失败。因此,采用三种不同的搜索连接策略,保证了对序列比对的结果,扩大的多序列比对的数据库范围,从而提升了多序列比对的质量;最后,使用了三种不同蛋白质单体多序列比对连接策略(基于基因距离,物种种类以及蛋白质交互网络),因此保证了对于任意一查询蛋白质复合物序列,都能产生其多序列比对结果。故本发明提升了模型的泛化能力。
- 还没有人留言评论。精彩留言会获得点赞!