基于神经网络的基因突变致病性检测方法、系统及介质与流程
本发明涉及的基因检测基因解读技术,具体涉及一种基于神经网络的基因突变致病性检测方法、系统及介质。
背景技术:
在基因检测中,如何从成千上万的基因突变中获得致病的基因突变,是基因解读的关键。传统的基因突变致病性分析的主要方法包括:(1)通过查找生物医学领域的专业数据库,看相应的基因或突变是否有致病的数据库记录,这些数据库包括omim、orphanet、hgmd、clinvar等;(2)通过蛋白质功能预测,判定该基因突变是否存在风险,常用的蛋白质功能预测软件如sift、polyphen2、mutationtaster、dann、cadd等;(3)根据该基因突变在人群中出现的频率,判定基因突变的风险,常用的人群数据库包括gnomad(genomeaggregationdatabase),exac(theexomeaggregationconsortium)和1kgp(1000genomesproject)等;(4)其他解读结果:如acmg(americancollegeofmedicalgeneticsandgenomics)突变解析、突变位点的保守性、突变是否在重复区域等等。因此,基因突变的分析结果涉及很多方面,结果数据繁多,单方面的分析结果都不能准确预测出基因突变的致病性强弱,专家需要综合分析这些项的结果,人工给出基因突变的致病性结果,这是一项十分耗时的工作。
如何从多方面的基因突变致病性分析结果得到一个致病性综合分析值,使得医生仅需关注致病性综合值较高的基因变异,大大减少医生所需关注的基因突变的数量,是基因解读需要解决的关键问题。目前这一领域最常用的方法是exomiser,它将基因突变从突变、算法和基因三个层面进行打分,再使用逻辑回归的方式对三种打分进行组合,获得一个致病性综合性分析值。这一方法最大的缺陷是考虑的致病性影响因素较少,而且有些因素(如老鼠和斑马鱼数据)对人的遗传病影响不大,有些关键性因素未考虑进去,导致获得的致病性综合性分析值准确性不高。
技术实现要素:
本发明要解决的技术问题:针对现有技术的上述问题,提供一种基于神经网络的基因突变致病性检测方法、系统及介质,与传统的人工解读和exomiser基因突变致病性综合性分析方法相比,本发明通过神经网络模型对训练样本进行学习,获取基因突变的大量特征和致病性之间的内在关联,不仅克服了人的主观性缺陷,而且能够综合考虑影响基因突变致病性的各种因素,使得综合分析的结果更为客观有效,从而大大提高了对基因突变致病性分析的准确性,提高了基因解读的效率。
为了解决上述技术问题,本发明采用的技术方案为:
一种基于神经网络的基因突变致病性检测方法,实施步骤包括:
1)输入待检测的基因检测vcf文件以及hpo表型;
2)根据待检测的基因检测vcf文件以及hpo表型获取各个基因变异的特征值;
3)对于每一种基因变异,将该基因变异的特征值输入训练好的神经网络模型得到该基因变异的致病性综合分析结果,所述神经网络模型被预先训练建立了各个基因变异的特征值、各个基因变异的致病性综合分析结果之间的映射关系;
4)按照致病性综合分析结果从高到低进行排序,确定每个基因变异的致病性优先级,按优先级顺序输出每个基因变异及其致病性综合分析结果。
可选地,步骤2)中各个基因变异的特征值包括:用于变异有无clinvar结果的特征;用于表示用户输入表型和当前变异基因对应表型的匹配度的特征genehpo;用于表示当前变异基因在omim数据库中是否有对应的疾病的特征inomim;用于表示当前变异基因在omim疾病库中对应疾病的hpo和用户输入的hpo是否相匹配的特征omimhpo;用于表示当前基因变异在多种人群数据库中出现的最大频率的特征maxmaf;用于表示蛋白质功能预测软件sift的预测结果的特征sift;用于表示蛋白质功能预测软件polyphen2预测结果的特征polyphen2;用于表示蛋白质功能预测软件mutationtaster预测结果的特征mtaster;用于表示保守性预测软件gerp++_rs结果的特征evorate;用于表示当前基因变异是否位于蛋白质的功能区域的特征domain;用于表示当前基因变异是否位于重复区域的特征rmsk;用于表示当前变异的接合性与相应基因对应疾病的遗传性是否一致的特征inherit。
可选地,步骤3)中的神经网络模型为bp神经网络。
可选地,所述bp神经网络为包括输入层、中间层以及输出层的三层结构,其中输入层节点数目与样本的特征数目相同,中间层节点数目大于输入层节点数目,输出层仅有一个节点。
可选地,步骤3)之前还包括训练神经网络模型的步骤,详细步骤包括:
s1)产生训练样本:(1)获取若干基因检测vcf文件、每个基因检测vcf文件对应的hpo表型和已知的致病基因;(2)获取基因突变的特征值:根据每个基因检测vcf文件以及hpo表型获取各个基因突变的特征值;(3)确定致病基因突变样本:对于每个基因检测vcf文件中致病基因所对应的所有突变,计算它们的突变注释评分p_score值,取突变注释评分p_score值最大的突变为致病基因突变,该基因突变为一个训练样本,并令其致病性综合分析值为第一标签值;(4)确定非致病基因突变样本:对于每个基因检测vcf文件,随机选取指定数量个非致病基因突变作为训练样本,并令其致病性综合分析值为第二标签值;
s2)训练神经网络模型:利用训练样本集完成神经网络模型的训练,使得神经网络模型被建立各个基因变异的特征值、各个基因变异的致病性综合分析结果之间的映射关系。
可选地,步骤s1)中突变注释评分p_score的计算函数表达式如下:
p_score=β1·protein+β2·(1-10*seg(maxmaf))+β3·clinvar/3
上式中,β1,β2,β3分别为三个权重参数,protein为蛋白质功能预测软件的最大预测值,seg(maxmaf)为特征maxmaf的转换结果,clinvar为用于变异有无clinvar结果的特征,蛋白质功能预测软件的最大预测值protein取用于表示蛋白质功能预测软件sift的预测结果的特征sift、用于表示蛋白质功能预测软件polyphen2预测结果的特征polyphen2、用于表示蛋白质功能预测软件mutationtaster预测结果的特征mtaster三者中的最大值,特征maxmaf的转换结果为在特征maxmaf小于等于预设门槛值时取原值否则取预设门槛值。
此外,本发明还提供一种基于神经网络的基因突变致病性检测系统,包括:
输入程序单元,用于输入待检测的基因检测vcf文件以及hpo表型;
特征值提取程序单元,用于根据待检测的基因检测vcf文件以及hpo表型获取各个基因变异的特征值;
致病性综合分析程序单元,用于对于每一种基因变异,将该基因变异的特征值输入训练好的神经网络模型得到该基因变异的致病性综合分析结果,所述神经网络模型被预先训练建立了各个基因变异的特征值、各个基因变异的致病性综合分析结果之间的映射关系。
此外,本发明还提供一种基于神经网络的基因突变致病性检测系统,包括计算机设备,该计算机设备被编程或配置以执行所述基于神经网络的基因突变致病性检测方法的步骤。
此外,本发明还提供一种基于神经网络的基因突变致病性检测系统,包括计算机设备,该计算机设备的存储器上存储有被编程或配置以执行所述基于神经网络的基因突变致病性检测方法的计算机程序。
此外,本发明还提供一种计算机可读存储介质,该计算机可读存储介质上存储有被编程或配置以执行所述基于神经网络的基因突变致病性检测方法的计算机程序。
和现有技术相比,本发明具有下述优点:与传统的人工解读和exomiser基因突变致病性综合性分析方法相比,本发明通过神经网络模型对训练样本进行学习,获取基因突变的大量特征和致病性之间的内在关联,不仅克服了人的主观性缺陷,而且能够综合考虑影响基因突变致病性的各种因素,使得综合分析的结果更为客观有效,从而大大提高了对基因突变致病性分析的准确性,提高了基因解读的效率。
附图说明
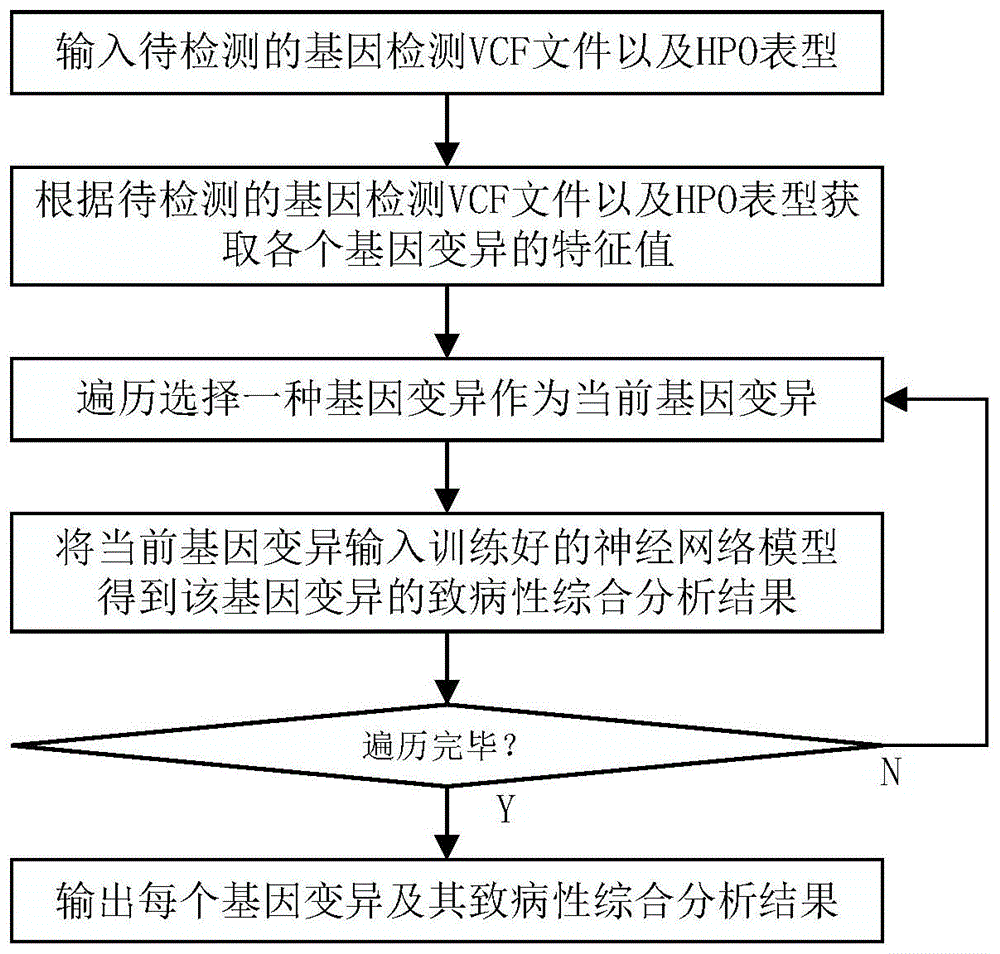
图1为本发明实施例方法的基本流程示意图。
具体实施方式
如图1所示,本实施例基于神经网络的基因突变致病性检测方法的实施步骤包括:
1)输入待检测的基因检测vcf文件以及hpo表型;
2)根据待检测的基因检测vcf文件以及hpo表型获取各个基因变异的特征值;
3)对于每一种基因变异,将该基因变异的特征值输入训练好的神经网络模型得到该基因变异的致病性综合分析结果,神经网络模型被预先训练建立了各个基因变异的特征值、各个基因变异的致病性综合分析结果之间的映射关系;
4)按照致病性综合分析结果从高到低进行排序,确定每个基因变异的致病性优先级,按优先级顺序输出每个基因变异及其致病性综合分析结果。
本实施例基于神经网络的基因突变致病性检测方法提出利用神经网络的方法进行基因突变的致病性分析,本实施例基于神经网络的基因突变致病性检测方法利用各种基因突变的致病性分析结果,构建若干神经网络的输入特征,并利用已知致病基因的基因检测vcf文件和hpo表型信息产生训练样本集,用于训练神经网络模型,将这些特征与基因变异的致病性之间的关系用网络权重表示出来,从而能够根据基因变异的特征,预测出基因突变致病性的综合分析结果。
步骤2)中根据基因检测vcf文件以及对应的hpo表型为输入,获取各个基因变异的特征值,这些特征值可在现有的工具软件的结果上计算获得,例如可在人和未来生物科技有限公司的gtx.digest软件(https://digest.gtxlab.com/)的注释结果的基础上计算获得。根据各种不同的致病性分析方法,本实施例步骤2)中各个基因变异的特征值包括:
1、用于该变异在clinvar数据库(http://www.clinvar.com/)有无相应结果的特征;无为0,有按致病等级取值为1,2,3,取值的确定方法如表1所示。
表1:用于变异有无clinvar结果的特征取值表。
2、用于表示用户输入表型和当前变异基因对应表型的匹配度的特征genehpo;genehpo的取值区间为[0,1]。特征genehpo的值按如下方式获取:令用户输入的hpo表型集合为ihpo;根据hpo数据集,检索与当前变异基因相关的hpo表型集合为ghpo,genehpo特征的取值按如下方式计算:
上式中,value(genehpo)表示特征genehpo的值,ghpo为hpo数据集中与当前变异基因相关的hpo表型集合,ihpo为用户输入的hpo表型集合,δ为参数,预设的一个很小的值,缺省为0.01,避免分母为零的情况,|a|表示集合a中元素的个数。
3、用于表示当前变异基因在omim数据库中是否有对应疾病的特征inomim;该特征的值为布尔值,如果有则为取值1,否则取值为0;
4、用于表示当前变异基因在omim疾病库中对应疾病的hpo和用户输入的hpo是否相匹配的特征omimhpo;该特征的值为布尔值,如果inomim中的取值为1,则获取这些omim疾病对应的hpo集合,如果该集合与用户输入的hpo集合有交集,则取值为1,否则取值为0;
5、用于表示当前基因变异在多种人群数据库中出现的最大频率的特征maxmaf;特征maxmaf的取值区间为[0,1],本实施例中选取六个与东亚人群密切相关的人群数据库,分别为exac_all,gnomad_exome_all,gnomad_exome_eas,1000g2015aug_eas,exac_eas,esp6500siv2_all,maxmaf的值为当前基因变异在这六个人群数据库中群体频率的最大值;
6、用于表示蛋白质功能预测软件sift预测结果的特征sift,取值规范到[0,1]区间,值越大表示变异的危害性越大;
7、用于表示蛋白质功能预测软件polyphen2预测结果的特征polyphen2,取值规范到[0,1]区间,值越大表示变异的危害性越大;
8、用于表示蛋白质功能预测软件mutationtaster预测结果的特征mtaster,取值规范到[0,1]区间,值越大表示变异的危害性越大;
9、用于表示保守性预测软件gerp++_rs结果的特征evorate;
10、用于表示当前基因变异是否位于蛋白质的功能区域的特征domain;特征domain的值为布尔值,如果是,则取值为1,否则取值为0;
11、用于表示表示当前基因变异是否位于重复区域的特征rmsk。特征rmsk的值为布尔值,如果是,则为1,否则取值为0。
12、用于表示当前变异的接合性与变异基因对应疾病的遗传性是否一致的特征inherit,如果相一致,则取值为1,否则取值为0;
本实施例中,步骤4)按照致病性综合分析结果从高到低进行排序以确定每个基因变异的致病性优先级的步骤,使得结果更加直观。
本实施例中,步骤3)中的神经网络模型为bp神经网络(backpropagationneuralnetwork,简称bpnn),此外也可以根据需要采用其他各类公知的神经网络模型。
本实施例中,bp神经网络为包括输入层、中间层以及输出层的三层结构,其中输入层节点数目与样本的特征数目相同,中间层节点数目大于输入层节点数目(缺省值为输入层节点数目的两倍),输出层仅有一个节点。
本实施例中,步骤3)之前还包括训练神经网络模型的步骤,详细步骤包括:
s1)产生训练样本:(1)获取若干基因检测vcf文件、每个基因检测vcf文件对应的hpo表型和已知的致病基因;(2)获取基因突变的特征值:根据每个基因检测vcf文件以及hpo表型获取各个基因突变的特征值;(3)确定致病基因突变样本:对于每个基因检测vcf文件中致病基因所对应的所有突变,计算它们的突变注释评分p_score值,取突变注释评分p_score值最大的突变为致病基因突变,将该基因突变入选为一个训练样本,并令其致病性综合分析值为第一标签值(本实施例中取值为1)。(4)确定非致病基因突变样本:对于每个基因检测vcf文件,随机选取2(可根据需要指定)个非致病基因突变作为训练样本,并令其致病性综合分析值为第二标签值(本实施例中取值为0);
s2)训练神经网络模型:利用训练样本集完成神经网络模型的训练,使得神经网络模型被建立各个基因变异的特征值、各个基因变异的致病性综合分析结果之间的映射关系。
本实施例中,步骤s1)中突变注释评分p_score的计算函数表达式如下:
p_score=β1·protein+β2·(1-10*seg(maxmaf))+β3·clinvar/3
上式中,β1,β2,β3分别为三个权重参数,protein为蛋白质功能预测软件的最大预测值,seg(maxmaf)为特征maxmaf的转换结果,clinvar为用于变异有无clinvar结果的特征,蛋白质功能预测软件的最大预测值protein取用于表示蛋白质功能预测软件sift的预测结果的特征sift、用于表示蛋白质功能预测软件polyphen2预测结果的特征polyphen2、用于表示蛋白质功能预测软件mutationtaster预测结果的特征mtaster三者中的最大值,特征maxmaf的转换结果为在特征maxmaf小于等于预设门槛值时取原值否则取预设门槛值。
其中三个权重参数β1,β2,β3分之和为1,即:
β1+β2+β3=1
本实施例中,默认取β1=0.2,β2=0.4,β3=0.4。
蛋白质预测值protein取sift,polyphen2,mtaster三个特征值中最大的一个,即:
protein=max(sift,polyphen2,mtaster)
seg(maxmaf)表示对特征值maxmaf进行如下变换:
即在特征maxmaf小于等于0.1时取原值,否则取预设门槛值0.1。
综上所述,本实施例提出了生成基因突变特征集的方法和产生训练样本集的方法,从而能够通过神经网络模型对训练样本集进行学习,获取特征和类别之间的内在关联,实现了利用神经网络进行基因突变致病性综合分析预测。该方法不仅克服了人工分析的主观性缺陷,而且能够综合考虑影响基因突变致病性的各种因素,使得综合分析的结果更为客观有效,大大提高了对基因突变致病性分析的准确性,提高了基因解读的效率。
此外,本实施例还提供一种基于神经网络的基因突变致病性检测系统,包括:
输入程序单元,用于输入待检测的基因检测vcf文件以及hpo表型;
特征值提取程序单元,用于根据待检测的基因检测vcf文件以及hpo表型获取各个基因变异的特征值;
致病性综合分析程序单元,用于对于每一种基因变异,将该基因变异的特征值输入训练好的神经网络模型得到该基因变异的致病性综合分析结果,神经网络模型被预先训练建立了各个基因变异的特征值、各个基因变异的致病性综合分析结果之间的映射关系。
此外,本实施例还提供一种基于神经网络的基因突变致病性检测系统,包括计算机设备,该计算机设备被编程或配置以执行前述基于神经网络的基因突变致病性检测方法的步骤。
此外,本实施例还提供一种基于神经网络的基因突变致病性检测系统,包括计算机设备,该计算机设备的存储器上存储有被编程或配置以执行前述基于神经网络的基因突变致病性检测方法的计算机程序。
此外,本实施例还提供一种计算机可读存储介质,该计算机可读存储介质上存储有被编程或配置以执行前述基于神经网络的基因突变致病性检测方法的计算机程序。
以上所述仅是本发明的优选实施方式,本发明的保护范围并不仅局限于上述实施例,凡属于本发明思路下的技术方案均属于本发明的保护范围。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理前提下的若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!