人CD133蛋白1-108肽段的新用途的制作方法
本发明涉及肽段的应用,尤其是人cd133蛋白1-108肽段(fecd133)在抑制cd133阳性肿瘤中的新用途,属于生物医药技术领域。
背景技术:
胶质瘤是最常见的原发性中枢神经系统肿瘤,约占所有颅内原发性肿瘤的一半。根据世界卫生组织(who)1999年的分类方案,可以分为神经母细胞瘤、星形细胞瘤、少支胶质瘤、室管膜瘤、混合型胶质瘤等。星形细胞瘤是神经胶质瘤中最常见的一类肿瘤,其中以胶质母细胞瘤恶性程度最高。每年约100000人中有5~6人被诊断为原发性的恶性脑肿瘤,其中约80%为恶性胶质瘤,其中有超过50%的案例为胶质母细胞瘤。而胶质母细胞瘤因为其高致残率、高死亡率引起了广泛的关注。尽管现在胶质母细胞瘤治疗手段丰富,手术切除伴随放化疗辅助治疗,其中位生存期仍只有12个月,攻克恶性胶质瘤一直是全球医疗期待解决的世纪难题。究其原因主要是因为胶质瘤肿瘤干细胞的存在,使胶质瘤在放化疗过程中极易发生药物耐受及转移。
人cd133最早是miraglia等1997年在造血干细胞/祖细胞表面发现,并认为是多种干/祖细胞的表面标志之一。该基因位于4号染色体,约152kb,属于膜蛋白超家族成员,是一个含有865个氨基酸,分子量为120kda,n端在胞外,5次跨膜的糖蛋白。而cd133作为肿瘤干细胞标志物的报导最早由singh等提出,他们以cd133作为标志物,应用免疫磁珠法从胶质瘤中分离出一小部分干细胞样细胞,在体外培养时表现出干细胞样表型,具体表现为:可以悬浮生长成神经球,并能不断的自我更新和增殖,同时在特定条件下如含血清的培养基中可以分化成与亲本肿瘤形态和分子表型相似的肿瘤细胞。随后,越来越多的研究发现cd133是一个广谱的肿瘤干细胞的生物标志物,如结直肠癌干细胞、肝癌干细胞、前列腺癌干细胞等。cd133阳性细胞远比cd133阴性细胞具有更强的成瘤、侵袭和耐药能力,肿瘤中cd133的高表达与肿瘤分期和更差的预后呈正相关。作为糖蛋白,人cd133胞外环有8个潜在的n-糖基化位点,n-糖基化影响细胞表面cd133的ac133抗原表位的识别,有助于cd133蛋白的稳定性,从而影响肿瘤的进程。
糖基转移酶在生物体内催化活化的糖链接到不同的受体分子,如蛋白、核酸、寡糖和脂上,糖基化的产物具有很多生物学功能并具有高度的底物专一性,参与体内重要的活性物质如糖蛋白和糖脂中糖链的合成,其作用是把相应的活性供体(通常是二磷酸核苷ndp-糖)的单糖部分转移到糖、蛋白质、脂类和核酸等,完成后者的糖基化加工,并实现其相应的生物功能。glt8d1即糖基转移酶8结构域1,其作为糖基转移酶的一种,比普通糖基转移酶更为复杂,其c-端结构增加识别未进行折叠的蛋白质(多肽)结构;glt8d1存在于人体绝大部分正常组织,包括脑组织。前期我们的研究发现,glt8d1是精神分裂症的风险基因,参与调控胚胎神经干细胞的增殖和分化能力,以及神经元的形态和突触传递等生理功能,通过影响神经发育,最终导致精神分裂症发生。
而glt8d1与cd133之间是否存在相互作用,如何相互作用,这种相互作用是否通过影响胶质瘤干细胞,对胶质瘤的发生发展起到了重要的作用,目前并无相关研究报道。
技术实现要素:
本发明的目的在于提供人cd133蛋白1-108肽段(fecd133)的新用途,即人cd133蛋白1-108肽段在制备抑制cd133阳性肿瘤的药物中的应用,所述人cd133蛋白1-108肽段的氨基酸序列如seqidno:1所示;人cd133蛋白1-108肽段作为药物能抑制cd133阳性肿瘤的增殖。
本发明还将人cd133蛋白1-108(fecd133)肽段应用在制备胶质瘤临床治疗药物中,人cd133蛋白1-108肽段的氨基酸序列如seqidno:1所示;
人fecd133是人cd133与人glt8d1蛋白相互作用的区段,并通过fecd133竞争结合glt8d1蛋白,阻断glt8d1蛋白对cd133蛋白的糖基化作用,使cd133蛋白不稳定而降解;从而有效降低胶质瘤干细胞活性,抑制胶质瘤细胞及胶质瘤干细胞增殖,促进肿瘤细胞及肿瘤干细胞凋亡,而肿瘤干细胞是肿瘤放化疗耐受和复发的原因。
本发明另一目的是将人cd133蛋白1-108肽段作为强化化疗药物应用在替莫唑胺化疗治疗胶质瘤中,人fecd133联合替莫唑胺应用于治疗胶质瘤,增强化疗效果。
所述fecd133蛋白序列为:
malvlgsllllglcgnsfsggqpsstdapkawnyelpatnyetqdshkagpigilfelvhiflyvvqprdfpedtlrkflqkayeskidydkpetvilglkivyyeag(seqidno:1);
我们通过免疫组化实验发现glt8d1蛋白在胶质瘤中高表达,并且与胶质瘤分级呈正相关,而敲低glt8d1的表达可以降低胶质瘤细胞克隆球的形成能力,提示glt8d1影响了胶质瘤干细胞的干性。进一步检测胶质瘤干细胞标志物发现肿瘤细胞的干性标志物表达显著下调,荧光共定位实验发现glt8d1与干细胞标志物cd133、sox2共定位;进一步在胶质瘤干细胞系gsc11、20171016b中敲低glt8d1的表达,抑制了胶质瘤干细胞的增殖,brdu的掺入显著下调,肿瘤微球形成能力显著下降,细胞凋亡明显增加。进一步的研究发现在胶质瘤干细胞中敲低glt8d1,其cd133阳性细胞率显著降低,其他干细胞标志物的蛋白水平也明显下降。glt8d1敲低的胶质瘤干细胞系在移植到裸鼠体内或原位注射后其成瘤能力也显著下降,小鼠生存时间更长。进一步的研究发现glt8d1可以与cd133直接相互作用,并糖基化cd133,维持cd133的蛋白稳定性,当cd133蛋白糖基化变弱的时候,cd133通过溶酶体途径降解。glt8d1的敲低使胶质瘤干细胞的干性下调,使细胞对替莫唑胺更加敏感。因此,我们利用截断体实验分析得到glt8d1和cd133的互作结构域,构建了仅表达cd1331-108个氨基酸的fecd133,发现其可以竞争阻断glt8d1和cd133的相互作用,降解内源的cd133,从而抑制胶质瘤干细胞的增殖,促进胶质瘤干细胞对替莫唑胺的敏感性。有意思的是fecd133对其他肿瘤来源的cd133阳性肿瘤干细胞具有同样的杀伤效果。
本发明明确了glt8d1的表达与胶质瘤干细胞干性标志物cd133之间的关系,并通过纯化的fecd133阻断glt8d1与cd133的相互作用,从而起到杀伤肿瘤的目的,而与替莫唑胺联合治疗胶质瘤的方式具有较大的应用价值和前景。
附图说明
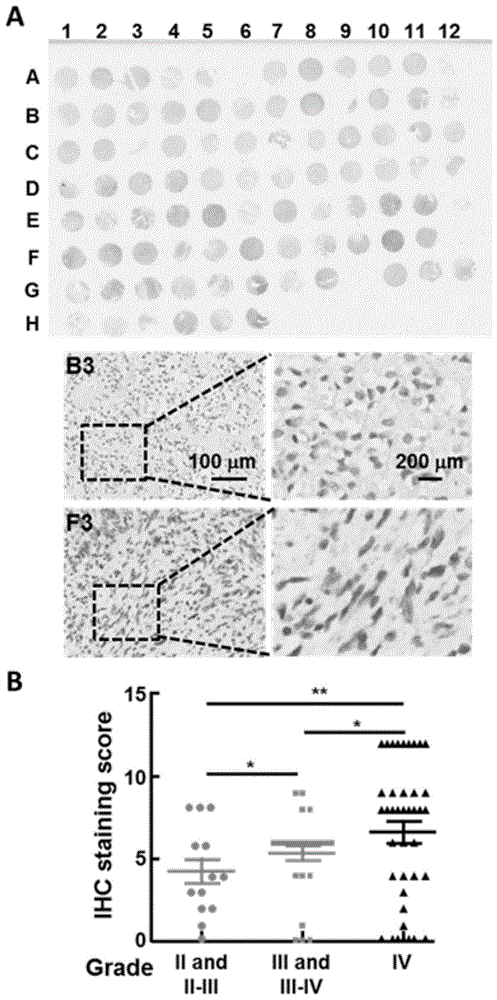
图1为人glt8d1在胶质瘤或正常组织中的表达结果,其中a图为免疫组织化学染色芯片全局图(上图)和局部放大图(下图)1-12表示横向有12个样本点,a-h表示纵向有8个样本点,共计90例;b图为人glt8d1基因表达与胶质瘤分级相关性结果;
图2为人glt8d1敲低或过表达对胶质瘤细胞克隆球形成能力的影响,其中a图为肿瘤克隆球形成情况展示图,b图为统计结果图;
图3为人glt8d1敲低或过表达对胶质瘤细胞肿瘤微球形成能力的影响,a图为u251和a172肿瘤微球示意图;b图为统计结果;
图4为人glt8d1敲低或过表达在极限稀释的情况下对胶质瘤细胞肿瘤微球形成能力的影响结果;
图5为人glt8d1敲低或过表达的细胞中胶质瘤干细胞标志物的蛋白表达情况;
图6为人glt8d1敲低或过表达的细胞中胶质瘤干细胞标志物的mrna表达情况;
图7为人肿瘤组织样本中glt8d1与cd133共定位的免疫荧光实验结果;
图8为人肿瘤组织样本中glt8d1、cd133与干细胞标志物sox2共定位;
图9为在胶质瘤干细胞系中构建glt8d1敲低或过表达稳转细胞系,左图为gsc11细胞系,右图为20171016b细胞系,其中上图(柱状图)为mrna表达结果;下图为蛋白表达结果;
图10为glt8d1敲低或过表达拯救敲低系的稳转胶质瘤干细胞细胞系的增殖情况,a图为gsc11细胞系,b图为20171016b细胞系;
图11为glt8d1敲低或过表达稳转胶质瘤干细胞细胞系的brdu掺入情况,a图为免疫荧光染色结果,b图为统计图;
图12为glt8d1敲低或过表达稳转胶质瘤干细胞细胞系的肿瘤微球形成情况,a图为肿瘤微球形成情况展示图,b图为统计图;
图13为glt8d1敲低或过表达稳转胶质瘤干细胞细胞系极限稀释条件下的肿瘤微球形成情况;
图14为glt8d1敲低稳转胶质瘤干细胞细胞系cd133的表达结果,左图为gsc11细胞系,右图为20171016b细胞系;
图15为glt8d1敲低稳转胶质瘤干细胞细胞系cd133阳性细胞率,左图为gsc11细胞系,右图为20171016b细胞系;
图16为构建的glt8d1敲低稳转胶质瘤干细胞细胞系中肿瘤干性标志物的表达情况;
图17为胶质瘤干细胞gsc11中敲低或过表达glt8d1的稳转细胞系按不同的数量接种到裸鼠体内后的移植瘤形成结果(极限稀释体内成瘤实验),a图为成瘤情况展示图,b图是敲低组(g8dsh#1,g8dsh#2)与对照组(ctrlshrna),过表达组(g8dove)与对照组(pcdh-vec)成瘤小鼠统计结果;
图18为glt8d1敲低稳转胶质瘤干细胞细胞系原位注射后成瘤及小鼠生存情况,a图为he染色结果展示图,b图为是敲低组(g8dsh#1,g8dsh#2)与对照组(ctrlshrna)小鼠的生存情况统计结果;
图19为glt8d1与cd133相互作用的免疫共沉淀实验结果,左图为gsc11细胞系中内源蛋白免疫共沉淀,右图为hek-293t细胞系中外源蛋白免疫共沉淀;
图20为glt8d1与cd133直接相互作用的结构域分析,a是不同肽段的表达图谱,b图是检测不同cd133肽段与glt8d1结合情况展示,c图是检测不同glt8d1肽段与cd133结合情况展示;
图21为敲低glt8d1降低cd133糖基化水平的结果示意图;
图22为glt8d1敲低导致的cd133蛋白降解加速;
图23为glt8d1敲低导致的cd133蛋白降解通路分析;
图24为cd133与溶酶体标志物cd63的共定位结果,a图为cd133与cd63共定位的染色示意图,b图为a图的统计结果;
图25为glt8d1敲低导致β-catenin入核受阻的结果示意图;
图26为glt8d1敲低导致wnt/β-catenin信号通路下游关键基因表达下调,而nh4cl处理可以阻断关键基因的下调表达的结果;
图27为glt8d1敲低细胞凋亡以及对替莫唑胺敏感性的结果;
图28为敲低glt8d1的u251细胞株及敲低稳转株中过表达cd133mrna的pcr检测结果(a图)和细胞增殖结果(b图);
图29为fecd133肽段的纯化及gst-fecd133pulldownglt8d1的结果;
图30为fecd133肽段处理的细胞中,cd133和β-catenin等的表达结果;
图31为fecd133肽段(右图)及替莫唑胺(左图)处理对正常胶质细胞和胶质瘤干细胞的杀伤结果;
图32为fecd133肽段对其他肿瘤cd133阳性和阴性细胞及正常细胞的的杀伤效果;左图为结直肠癌,右图为肝癌细胞;
图33为fecd133肽段对β-catenin信号通路关键分子β-catenin入核的影响结果;
图34为fecd133肽段对β-catenin信号通路下游通路相关基因mrna表达的影响结果;
图35为fecd133肽段与替莫唑胺协同促进胶质瘤干细胞的凋亡;a图为流式细胞分析展示图,b图为统计结果;
图36为fecd133肽段和/或替莫唑胺处理胶质瘤干细胞gsc11时,细胞内凋亡相关蛋白的检测;
图37为fecd133肽段和/或替莫唑胺处理裸鼠移植瘤的作用结果,a图裸鼠移植瘤大小示意图,b图肿瘤重量的统计结果,c图是a图中肿瘤体积的统计结果;
图38为fecd133肽段和/或替莫唑胺处理原位移植瘤对体内原位移植瘤的抑制作用结果。
具体实施方式
下面通过实施例对本发明作进一步详细说明,但本发明的保护范围不局限于所述内容,实施例中方法如无特殊说明均采用常规方法,使用试剂如无特殊说明,均为常规市售试剂或采用常规方法配置的试剂。
实施例1:病理生理学检测
胶质母细胞瘤芯片委托上海芯超公司实施免疫组化实验,分析glt8d1的高低表达与胶质瘤病人的临床分级的相关性;对实验动物体内取出的肿瘤组织实施he染色实验;
免疫组织化学染色实验如下:4%pfa将待检测组织固定过夜,用流水冲洗已经固定的组织块30min;75%乙醇1h;80%乙醇1h;90%乙醇1h;95%乙醇ⅰ1h;95%乙醇ⅱ1h;100%乙醇ⅰ1h;100%乙醇ⅱ1h;二甲苯ⅰ35min;二甲苯ⅱ35min;浸蜡:石蜡ⅰ1h;石蜡ⅱ1h;将组织包埋进模具中备用;切片前首先将石蜡块-20℃冰冻15min,切片3μm厚度,挑选完整平整的切片在56℃热水中展片,65℃烘片30min;然后进行染色操作:二甲苯3缸,每缸10min;无水乙醇2缸,每缸2min;95%乙醇2min;85%乙醇2min;75%乙醇2min,流动自来水冲洗2min;蒸馏水洗2min;样品放入柠檬酸修复液中,用高压灭菌锅进行抗原修复25min,拔掉电源10min,打开高压锅盖子,将盛有柠檬酸抗原修复液(含样品)的容器取出水浴至常温;将样品用蒸馏水洗1min;3%双氧水孵育20min;油性笔将样品画在圈内,pbs+1%tween-20+10%正常山羊血清滴在圈内样品表面,封闭半小时;孵育一抗glt8d1过夜;pbs清洗3min,重复两次;pbs+1%tween-203min;孵育二抗1h,pbs冲洗3min,重复两次;pbs+1%tween-20,3min;dab显色5min;蒸馏水洗1min;苏木素2-3min;蒸馏水洗1min;1%盐酸酒精分化数秒;蒸馏水蓝化2min;75%乙醇20s;85%乙醇20s;95%乙醇30s;无水乙醇2缸,每缸30s;二甲苯2min,重复两次;封片;
he染色如下:准备好的切片进行二甲苯ⅰ处理5min;二甲苯ⅱ处理5min;100%乙醇2min;95%乙醇2min;85%乙醇2min;75%乙醇2min;自来水洗2min;蒸馏水洗2min;苏木素5-10min;自来水洗1min;1%盐酸酒精分化数秒(眼观淡紫红色为度);自来水洗1min;温水(50℃)蓝化5min;或用1%氨水蓝化30s,自来水洗5-10min;蒸馏水1min;95%乙醇1min;伊红数秒。系列梯度酒精脱水和透明:75%乙醇20s;85%乙醇20s;95%乙醇30s;95%乙醇30s;100%乙醇30s;100%乙醇30s。二甲苯2min;二甲苯2min;最后二甲苯没干时进行封片。
结果如图1所示,在胶质瘤组织中,人glt8d1的表达显著高于正常组织,且其表达量的高低与胶质瘤恶性程度呈正相关,分级越高,glt8d1的蛋白表达越高;ⅲ及ⅲ-ⅳ期的蛋白表达比ⅱ及ⅱ-ⅲ的高,ⅳ的蛋白表达比ⅲ及ⅲ-ⅳ期的高。
实施例2:人glt8d1敲低或过表达对胶质瘤细胞克隆球形成影响实验
1、构建glt8d1稳定敲低或过表达的细胞系
利用慢病毒基因过表达载体pcdh-mscv-egfp-3×flag,构建glt8d1过表达载体,设计pcr引物扩增人glt8d1基因的编码区序列,并克隆到上述过表达载体中,pcr引物为f:atgtcattccgtaaagtaaac,r:tcactttatgtttgagatctc,阴性对照为pcdh-mscv-egfp-3×flag;利用plko.1shrna表达载体,根据人glt8d1基因序列设计shrna靶序列,合成2对寡核苷酸序列,同时合成对照寡核苷酸序列,将以上寡核苷酸序列耦合后克隆到plko.1载体上,阴性对照scrambleshrna的序列为:gcactaccagagctaactcag;21bp靶向glt8d1的shrna序列分别为:shrna#1:agccagcacttgctcatttaa;shrna#2:tctcaggaagtcctggaagat。在肿瘤细胞系u251、a172中利用hek-293t细胞和包装质粒包装慢病毒,收集产生慢病毒颗粒的培养基上清并感染目的细胞,通过载体上含有的耐药性标记嘌呤霉素,进行筛选而得到稳定敲低或过表达细胞株;胶质瘤干细胞系gsc11和20171016b的glt8d1稳定敲低或过表达细胞株的构建方法同上,如图9所示显示,无论是rna水平还是蛋白水平,胶质瘤干细胞系稳转细胞株构建成功;
2、先配制1.2%和0.6%的agar胶高压蒸汽灭菌锅中灭菌,放烘箱备用;将1.2%琼脂糖胶与经37℃预热的20%fbs的细胞培养液按体积比1:1完全混合后边晃动边加到六孔板中;20min后待下层胶完全凝固,取3×103/ml单细胞悬液(20%fbs)与0.6%胶1:1混匀,按照每孔2ml接种进六孔板,即每孔3×103细胞;待上层胶凝固之后,补加细胞培养液每孔2ml;隔天换液至形成克隆球后,小心地去除上清,用4%多聚甲醛室温固定1h后,用0.05%的结晶紫染色后拍照计数;
结果如图2所示敲低glt8d1的胶质瘤细胞(u251和a172)的克隆球形成能力减弱,而过表达则反之;
3、取对数期生长的glt8d1稳定敲低或过表达细胞株,消化制成单细胞悬液,进行浓度测定;按照每孔3×104个细胞接种于超低吸附的6孔板中,并设置3个生物学重复孔,每3天补加20%的培养基,10-14天后计数形成的肿瘤微球个数;
结果如图3所示,敲低了人glt8d1的胶质瘤细胞(u251和a172),肿瘤微球形成能力变弱,而过表达则反之;
图12的结果显示胶质瘤干细胞(gsc11和20171016b)构建的稳转细胞株的肿瘤微球的形成能力与胶质瘤细胞(图3)中表现一致。
4、极限稀释实验
取对数期生长的glt8d1稳定敲低或过表达细胞株,消化制成单细胞悬液进行浓度测定;按照每孔5、10、20、50、100个细胞的梯度接种于超低吸附的96孔板中,并设置5个生物学重复孔,10-14天后计数形成的肿瘤微球个数;
结果如图4所示,u251中敲低了glt8d1,肿瘤微球形成能力变弱,而过表达则反之,接种的细胞数量无论多少,从每孔接种的10个细胞到100个细胞都表现出了相同的趋势。
图13结果显示在胶质瘤干细胞系gsc11中进行的极限稀释肿瘤微球形成实验,其结果显示与图4结果一致。
实施例3:glt8d1敲低或过表达的细胞中胶质瘤干细胞标志物免疫印迹检测实验
按照特定实验处理后的细胞,弃去上清培养基,用pbs洗涤1次;根据细胞沉淀的量加入相应的细胞裂解液,于冰上反复冻融裂解3次,期间不停吹打;4℃、15000rpm/min离心10min,取上清弃沉淀用于后续实验。bca蛋白定量后加入5×上样缓冲液,置于100℃金属干热仪中煮5min;直接进行聚丙烯酰氨凝胶(sds-page)电泳,或者取出后置于冰上冷却分装,-80℃保存。每孔加入蛋白量30-50μg,首先80v电压下电泳跑完浓缩胶,再用120v电压电泳直至溴酚蓝跑出。转膜:把转膜架放在转膜液中,黑色塑料板面向下,白色在上,在黑色面上方放一块海绵垫,海绵垫上方放两张滤纸;轻轻把凝胶从电泳玻璃板上取下来后放到滤纸上,把pvdf膜放到凝胶上方,再放两张滤纸,滤纸上方放一块海绵垫,把黑白两块塑料板固定牢固,放入已经预冷的转膜缓冲液中;冰浴条件下,83v电压转膜3h;把pvdf膜放入含5%脱脂奶粉的tbst中,在摇床上缓慢摇动,室温封闭2h;加入用含3%bsa的tbst稀释的一抗,4℃缓慢摇动中孵育过夜;tbst洗膜10min×3次;加入hrp标记的二抗室温孵育2h;tbst洗膜10min×3次;把pvdf膜转移到发光板上,避光条件下加入ecl试剂(用前把a和b液进行等量混合),显影拍照。
结果如图5所示,在敲低glt8d1的情况下,胶质瘤干细胞标志物如cd133、β-catenin、sox2、oct4等都显著下调,而过表达,则这些干细胞标志物的表达显著上升;
图9下图的结果显示在胶质瘤干细胞系(gsc11和20171016b)中敲低或过表达的稳转细胞株成功构建,敲低组的细胞株中蛋白表达较对照组(ctrlshrna)显著下调,过表达的细胞株(g8dove)中myc-glt8d1的表达受到诱导;
图16的结果显示敲低glt8d1的表达,胶质瘤干细胞的干细胞标志物如cd133、β-catenin、sox2、oct4等都显著下调;
图22的结果显示在放线菌酮(cycloheximide,chx)处理的情况下,当glt8d1敲低时,随着时间的推移,cd133蛋白的半衰期显著较对照组短。
图30的结果表明在胶质瘤干细胞中,用fecd133处理能降低内源cd133的蛋白稳定性和β-catenin的蛋白表达,cd133、β-catenin的表达显著降低;
图36的结果显示,在胶质瘤干细胞中用不同如下方式进行处理:对照组(gst)、替莫唑胺(tmz)、fecd133、替莫唑胺联合fecd133处理,细胞内凋亡相关蛋白的表达情况,替莫唑胺联合fecd133处理凋亡相关蛋白:cleavedparp(cparp)、cleavedcaspase3(cc3)、bax的表达显著上调,而抑制凋亡的蛋白bcl-2则显著下调。
实施例4:反转录和实时定量pcr
取1μg提取的rna进行反转录。具体如下:去除基因组dna反应,按如下成分于冰上配制反应混合液,然后再分装到每个反应管中,最后加入rna样品;轻柔混匀,按照42℃,2min进行反应;
反应后样品置于冰上,先按反应数+1的量配制混合mix,然后每个反应管分装10μl:
轻柔混匀后立即进行反转录反应:反应程序为37℃,15min;85℃,5s,4℃;
每个样本设置三个重复孔,按如下组分配制:
混匀以上组分,加入到96孔板各孔,封膜后,离心使液体聚集到管底;根据以下条件进行pcr反应,热循环参数如下:50℃,2min;95℃,2min;95℃,10min;95℃,15s,60℃,1min,40个循环;
相关扩增引物序列如下:
β-actin–f:aagtgtgacgtggacatccgc;β-actin–r:ccggactcgtcatactcctgct;
glt8d1-f:acttgccaattctggttccca;glt8d1-r:cggatgacaactttagtagaggc
cd133-f:atggcaacagcgatcaagg;cd133-r:gtactttgttggtgcaagctct;
axin2-f:agtcagcagagggacaggaa;axin2-r:cttcgtacatggggagcact;
c-myc-f:ggctcctggcaaaaggtca;c-myc-r:ctgcgtagttgtgctgatgt;
sox2-f:cacagatgcaaccgatgca;sox2-r:ggtgccctgctgcgagta;
cd44-f:ctgccgctttgcaggtgta;cd44-r:cattgtgggcaaggtgctatt;
结果如图6所示,cd133的rna水平在敲低或过表达的时候都没有显著变化,而其他的干细胞标志物sox2、oligo2、oct4及cd44的rna水平与glt8d1的表达水平成正相关,即glt8d1低表达,则sox2、oligo2、oct4及cd44的rna表达水平低,而glt8d1高表达,则sox2、oligo2、oct4及cd44的rna表达水平高。
图9上图(柱状图)glt8d1的mrna表达水平也显示构建的胶质瘤干细胞(gsc11和20171016b)中敲低组(g8dsh#1,g8dsh#2)glt8d1的mrna表达水平较对照组(ctrlshrna)显著降低,而过表达组(g8dove)glt8d1的mrna表达水平较对照组(pcdh-vec)显著升高;
图26的结果显示敲低glt8d1,c-myc、axin2和cyclind1的rna表达水平显著降低,而这一表型可以被nh4cl逆转;
图28a的结果显示在敲低glt8d1的u251细胞系及敲低稳转系中过表达cd133的细胞表达glt8d1和cd133的情况,证明了细胞系构建成功。
图34的结果显示在不同浓度的fecd133的处理下,axin2和c-myc的mrna随着加入fecd133肽的浓度的增加而降低。
实施例5:免疫荧光实验
取对数期生长的细胞系,消化制成单细胞悬液,进行浓度测定;按照每孔3×104个细胞接种于8孔板中,培养24h后,取出用4%多聚甲醛室温固定20min,加pbs+0.3%tween-20洗去残余多聚甲醛,加pbs+0.3%tritonx-100室温处理3min破膜,后用pbs+0.3%tween-20+10%正常山羊血清室温封闭30min,pbs+0.3%tween-20+5%正常山羊血清配置一抗,4℃孵育一抗过夜,第二天取出8孔板,用pbs+0.3%tween-20洗去残余一抗,洗3次,每次10min,用pbs+0.3%tween20+5%正常山羊血清配置二抗并室温孵育二抗2h后用pbs+0.3%tween-20洗去残余一抗,洗3次,每次10min,封片拍照;
图7结果表明在病人的肿瘤组织样本中,人glt8d1与cd133存在共定位;
图8结果表明glt8d1与cd133共定位的细胞类型是sox2阳性的胶质瘤干细胞;
图24结果显示cd133能与溶酶体标志物cd63共定位,且在敲低glt8d1的细胞中,cd133与cd63共定位的细胞数显著上升,表明cd133通过溶酶体途径降解;
图25结果显示glt8d1敲低能抑制β-catenin入核;在敲低glt8d1的稳转细胞(g8dsh#1,g8dsh#2)中,β-catenin入核显著较对照组细胞(ctrlshrna)少。
实施例6:细胞增殖实验
取处于对数期生长的稳定细胞系,用0.25%的胰酶消化制成单细胞悬液,进行浓度测定;按照所需细胞数gsc11:1.5×104/孔、20171016b:1.5×104/孔、u251:1.5×104/孔、a172:1.5×104/孔,用完全培养基配制相应的细胞悬液种至12孔板中(1.0ml/孔),37℃、5%co2培养6天;每天同一时间进行计数,绘制出生长曲线;
如图10所示,在胶质瘤干细胞系(gsc11、20171016b)中,敲低glt8d1(g8dsh#1,g8dsh#2)的细胞系,细胞的生长明显放缓,而在敲低株中过表达glt8d1(g8dsh#2+g8dove),可以加速细胞的生长,拯救因glt8d1的低表达量造成的生长缓慢;
图28b的结果显示了在敲低glt8d1的u251细胞系及敲低稳转系中过表达cd133的细胞增殖情况,过表达cd133可以拯救因敲低glt8d1造成的生长缓慢。
实施例7:brdu掺入实验
取处于对数期生长的稳定细胞株,用0.05%的胰酶消化制成单细胞悬液,进行浓度测定按照每个孔3.5×104细胞总量种到8孔板中。再将brdu(终浓度为10μm)加入细胞培养基中,37℃孵育20min,取出后用pbs轻轻地润洗一次,弃去上清,用4%多聚甲醛室温固定20min,后用pbs洗1次,随后加入2nhcl-0.5%trionx-100室温孵育30min破细胞核膜,用1mnahco3中和至无泡沫产生后用pbs+0.1%tween-20洗两次,用10%正常山羊血清室温孵育半小时,用pbs+0.1%tween-20洗两次,加入brdu(1:1000使用)一抗,然后4℃孵育过夜。用pbs+0.1%tween-20清洗3次,每次10min,加入荧光二抗(1:1000使用)和dapi(1:1000使用),避光室温孵育2h,弃去未结合的染料,pbs+0.1%tween-20清洗三次,每次10min,后加入pbs+0.1%tween-20避光保存,显微镜观察、拍照后计数;
图11的结果显示稳转的glt8d1敲低细胞系(g8dsh#1,g8dsh#2)中brdu的掺入明显较对照组(ctrlshrna)减少,过表达组(g8dove)brdu的掺入水平较对照组(pcdh-vec)显著升高。
实施例8:流式检测细胞cd133表达
将gsc11和20171016b的对照和稳定敲低细胞分别种于6孔板中,每孔3×105个细胞;隔天,待细胞生长状态良好后,用0.05%的胰酶消化3min成单个细胞悬液,离心去上清,加pbs+1%bsa洗两次,洗去残余的胰酶。加500μl的pbs和5μl的pe-cd133抗体,另取一管对应的细胞加入5μl的pe-mouse抗体作为阴性对照;上机检测,分析结果;
图14、15结果显示与对照组(ctrlshrna)相比,glt8d1敲低的稳转细胞内的cd133表达率显著降低。
实施例9:细胞凋亡检测
u251、a172、gsc11和20171016b稳定敲降细胞生长到80%覆盖度时,去掉培养基上清,pbs清洗一次,胰酶1ml消化3min,然后加入5ml培养基终止制成单细胞悬液后计数,按照每孔2ml培养基体积,细胞总量4×105-6×105个,分别种于6厘米培养皿中,每个样品设置三个重复孔,轻轻混匀以保证细胞分布均匀,然后置于37℃、5%co2二氧化碳培养箱中过夜。24h后,加入药物(替莫唑胺tmz:200μm;fecd133:40nm;gst:40nm)处理48h,收集培养基上清,以收集漂浮在上清里的凋亡的细胞;pbs清洗一次,胰酶0.5ml消化3min,然后加入2ml培养基终止,吹打成单细胞悬液,注意轻柔吹打,防止因机械力带来的细胞损伤凋亡,收集细胞到离心管中,2000rpm/min离心5min,去掉上清液,加入5mlpbs洗一次,2000rpm/min离心5min,重复一次;按照annexinv-apc/pi细胞凋亡检测试剂盒说明书,先加入1×bindingbuffer将细胞重悬,取出三个对照所需细胞(阴性对照,apc单标对照,pi单标对照),然后每管样品加入apc和pi各5μl到细胞悬液中(对照样品为:不加染料,只加5μlapc,只加5μlpi),轻柔混匀;37℃避光孵育30min;利用流式细胞仪检测细胞凋亡比例的变化;数据进行整理分析,绘制细胞凋亡分布图和统计图;
图27的结果显示在glt8d1敲低的细胞中,细胞凋亡显著增加,而当glt8d1敲低的细胞用替莫唑胺(tmz)处理后,细胞凋亡进一步增加;
在胶质瘤干细胞系中进行不同的处理:对照组(gst)、替莫唑胺(tmz)、fecd133、替莫唑胺联合fecd133处理,图35的细胞内凋亡情况流式分析结果显示:fecd133和替莫唑胺协同处理>fecd133处理>替莫唑胺处理>gst对照。
实施例10:体内极限稀释裸鼠成瘤模型
将105、104、103、102个的gsc11稳转敲低和对照肿瘤细胞分别注射至5~6周龄的balb/c(nu/nu)裸鼠腋窝部位,实时监测接种部位的肿瘤的重量、肿块的体积等,绘制曲线;三个月后,待肿瘤体积长到一定比例,将裸鼠处死并解剖出肿块,记录和拍照;
图17是在极限稀释条件下,胶质瘤干细胞gsc11中敲低或过表达glt8d1的稳转细胞系按不同的数量接种到裸鼠体内后的移植瘤形成结果,结果显示敲低组的移植瘤成瘤能力显著下降,而过表达组则相反。
实施例11:颅脑原位注射成瘤模型
取5周大nod/scid雌鼠,将生长到70%丰度左右的稳转敲低和对照gsc11细胞;去培养基加1ml0.05%的胰酶消化3min,计数得总细胞数,离心去培养基,加一定量的pbs重悬细胞使之浓度为1.67x108个/ml;注射3μl细胞悬液,即5×105的细胞。将小鼠按照10μl/g的剂量麻醉后将小鼠固定于脑损伤仪,耳杆插入小鼠耳朵,两端保持同等距离,注意避开迷走神经,用碘酒擦拭脑部消毒,用手术刀在脑中线划开头部皮肤。在左脑矢状缝上方贴近中线的位置用牙钻钻开1-2mm直径的正方形,去除颅骨,用棉签拭去血迹,调节注射泵速度为5nl/s,泵入细胞悬液3μl(5×105个细胞),移动注射泵定位0点,记录坐标;根据需要调节坐标位置(x:+2.5mm;y:-1mm;z:-3.5mm),将细胞悬液泵入颅内,泵入细胞后,停留5-10min,然后缓慢旋出针头,旋出时间不能小于10min,以上完成后,将耳杆拿掉,从固定仪上取下小鼠,用棉签擦去血迹,用镊子整理头部皮肤,将其缝合涂上青霉素软膏,将小鼠置于电热毯上,使其恢复正常,定期观察小鼠状态。待小鼠生长状态较差至即将死亡时,取出小鼠全部脑子,放入4%的多聚甲醛4℃过夜固定,后脱水包埋,切片并实施he染色观察肿瘤的形成大小;
图18是胶质瘤干细胞gsc11中敲低glt8d1的稳转细胞系原位注射接种到裸鼠颅脑中的成瘤实验结果,在敲低glt8d1后胶质瘤干细胞原位注射成瘤能力显著低于对照组,小鼠的生存时间亦较对照组显著延长,即敲低组(g8dsh#1,g8dsh#2)较对照组(ctrlshrna)生存时间显著延长。
实施例12:蛋白相互作用检测
免疫共沉淀包含了内源免疫共沉淀和外源免疫共沉淀;内源免疫共沉淀:用ipbuffer(20mmtris-hcl(ph7.4)、1%tritonx-100、0.1%sds、150mmnacl、0.5mmedta)裂解液裂解处于对数生长期的gsc11细胞,冰上放置0.5h,期间用液氮对该样品进行反复冻融处理,15,000rpm/min离心10min,取上清,并将上清分为3管,每管500μl,加入适量的glt8d1,cd133和阴性对照兔igg抗体。4℃摇床中孵育过夜。第二天加入proteingbeads30μl4℃摇床摇2h,离心去上清,加ipbuffer500μl,4℃摇床摇5min洗去残余蛋白,重复操作3次;加5×上样缓冲液,置于100℃金属干热仪中煮5min后蛋白电泳;
外源免疫共沉淀:瞬时转染pcdna3.1-myc-glt8d1和lv-cd133-flag各12.5μg的质粒到hek293t细胞中,8h后更换新鲜培养基,48h后收集细胞沉淀,并将细胞沉淀融于适量ipbuffer中,加入myc(santacruz,1:500)或flag(sigma,1:500)的的标签抗体,4℃摇床中孵育过夜;第二天加入proteingbeads30μl4℃摇床摇2h,离心去上清,加ipbuffer500μl,4℃摇床摇5min洗去残余蛋白,重复操作3次。加5×上样缓冲液,置于100℃金属干热仪中煮5min后蛋白电泳。
图19结果显示无论是在胶质瘤干细胞系gsc11(内源免疫共沉淀),还是在hek-293t细胞(外源免疫共沉淀)中,免疫共沉淀实验都显示glt8d1和cd133之间存在相互作用;
图20结果显示将glt8d1和cd133截成不同大小的肽段构建质粒,共转入hek-293t细胞(外源免疫共沉淀)进行相互作用结构域分析,a是不同肽段的表达图谱,b图是检测不同cd133肽段与glt8d1结合情况分析,发现c3即1-108位cd133的肽段是cd133与glt8d1的结合的关键区域,c图是检测不同glt8d1肽段与cd133结合情况分析,发现g3即29-220位glt8d1的肽段是glt8d1与cd133结合的关键区域。
实施例13:gstpull-down实验
将cd133的蛋白编码区序列克隆到pgex-4t-1的载体中,用pgex-4t-1空载作为对照;取500ngpgex-4t-1-cd133的质粒加到bl21(de3)的感受态中,冰上放置30min,42℃热激90s,冰上放置5min,后涂板。37℃培养过夜;挑单克隆到5ml的含有氨苄抗性的培养基中,37℃培养至od600值为0.8-1.0之间;取过夜培养的菌1:100加到500ml含有氨苄抗性的培养基中。37℃培养至od600值为0.7后用0.3μm的iptg过夜诱导16h。第二天将过夜诱导的菌以5000rpm/min的速度4℃离心10min,去培养基,用50ml预冷的pbs洗去残余培养基重悬细菌。5000rpm/min的速度4℃离心10min;加30ml预冷pbs和1mm的dtt,100mm的pmsf(蛋白酶抑制剂)。超声破碎细胞。取1l烧杯放满冰,将待超声的细胞放在50ml的离心管中,插在漂浮板上超声破碎(超声2s,停2s,共80次),超声后1:100加tritonx-100,冰上30min,继续裂解细胞。12,000rpm/min,4℃,离心30min。将gstbeads混匀后加100μl的gstbeads到柱子中,加10ml的pbs洗beads,让其缓慢流淌,重复加10ml的pbs洗beads,让其缓慢流淌,全程不能让柱子干燥。将离心后的融合蛋白样品用枪头引流缓慢过柱;柱子底下用50ml离心管中接住留下来的flowthrough,将flowthrough重新过柱2次;用10mlpbs-1%tritonx-100低盐缓冲液洗beads之后再用10ml含1mnacl的pbs-1%tritonx-100高盐缓冲液洗beads,最后再用10mlpbs-1%tritonx-100低盐缓冲液洗beads;取一部分beads制样检测纯化的gst融合蛋白的情况,封住柱子底部,加入用ipbuffer裂解好的gsc11细胞样品与纯化的融合蛋白孵育过夜;第二天,经低盐、高盐、低盐缓冲液充分洗涤之后,用elutebuffer反复洗脱3次;后将洗脱后的蛋白样品做免疫印迹实验,观察glt8d1蛋白与cd133蛋白是否相互作用;
结果如图29,用gst进行的pulldown样品中没有检测到glt8d1,而gst-fecd133进行的pulldown样品中检测到glt8d1,表明glt8d1蛋白可以与fecd133直接相互作用。
实施例14:考马斯亮蓝染色方法
将实施例13中的样品跑完的聚丙烯酰胺凝胶用去离子水反复冲洗胶板,小心的撬开薄板,并将凝胶小心的置于装有50ml的考马斯亮蓝染色液的15cm培养皿中,并放于室温摇床染色1h,回收考马斯亮蓝染色液并加入50ml的脱色液,每隔30min更换一次脱色液,脱色3h左右即可,取出凝胶拍照;
结果如图29下图,通过考马斯亮蓝染色检测结果显示纯化的gst和gst-fecd133蛋白的纯度很高。
实施例15:糖基化修饰
用ripa裂解液分别裂解glt8d1的稳定敲低组与对照组细胞样品,并做蛋白免疫印迹实验,观察敲低组蛋白样品中cd133蛋白的分子量与对照组样品比是否有偏移;
图21的结果显示在hek-293t单转glt8d1(敲低和对照)质粒,cd133蛋白的分子量下调,与swainonine处理的情况类似,蛋白稳定性也有显著的下调,而当glt8d1相关质粒与cd133糖基化缺陷型共转时,cd133等的蛋白分子量进一步下调,与pngasef处理的情况一致,swainonine能抑制n-连接糖基化,从而阻止天冬酰胺-连接的糖蛋白合成,可逆抑制溶酶体α-甘露糖苷酶和高尔基体α-甘露糖苷酶ii活性,中断高甘露寡糖形成复杂型寡糖的过程;而pngase酶能去除高甘露糖型、复合型、杂合型糖链,去糖基化更彻底。表明glt8d1可以n-连接糖基化cd133,当cd133蛋白糖基化受到抑制时,蛋白稳定性变弱。
实施例16:蛋白降解分析实验
将生长状态良好的待检测细胞,接种于6孔板中,置于37℃、5%co2二氧化碳培养箱中过夜;第二天,待细胞生长状态良好后,分别加入50mm的nh4cl和20μm的mg132,dmso被用来作为阴性对照,药物处理24h后,收集细胞并加ripa裂解液裂解细胞用于蛋白免疫印迹分析;
图23的结果说明在溶酶体降解途径抑制剂氯化铵(nh4cl)处理的情况下,cd133蛋白的稳定性显著增加,而对照组泛素蛋白酶体抑制剂mg132处理则与对照无显著区别,即glt8d1敲低导致的cd133蛋白降解可以被nh4cl阻断,而不被mg132阻断,表明cd133的降解是通过溶酶体降解途径。
实施例17:核质分离实验
将待检测的生长状态良好的细胞传代至10cm培养皿中,然后置于37℃、5%co2二氧化碳培养箱中过夜培养。第二天,待细胞生长状态良好后,加入胰酶消化成单个细胞并离心得到细胞沉淀。然后加入1.5ml的buffera,用移液枪反复吹打并确保裂解充分,并放置于冰上冰浴5min;4℃离心机2700rpm/min离心4min,小心地取上清转移至新的ep管中,该上清即为胞质蛋白。用1ml的buffera洗3次沉淀,该沉淀即为细胞核;加入3倍沉淀体积的bufferb,用枪头反复吹打,并置于冰上30min冰浴,以确保裂解充分;最后将胞质蛋白和核蛋白分别15,000rpm/min离心10min。将上清转移至新的ep管中,用于蛋白印迹分析;buffera:20%甘油、10mmnacl、1.5mmmgcl2、5mmedta(ph8.0)、20mmtris(ph7.5)、0.1%np40;bufferb:10%甘油、100mmnacl、1.5mmmgcl2、5mmedta(ph8.0)、20mmtris(ph7.5),1%np40;
图33核质分离的结果显示,fecd133肽段处理的胶质瘤干细胞,其β-catenin蛋白的入核受到显著抑制。
实施例18:体外药敏实验
当目的细胞生长至密度约为70%-80%左右时,使用胰酶将细胞消化成单细胞悬液,按照每孔约8000-10000个细胞100μl体积将其种在96孔板中。细胞贴壁24h后,此时目的细胞密度约为50%。轻轻地吸取孔中的培养基弃去,并沿壁缓缓地加入含fecd133或tmz的新鲜培养基,终体积为每孔100μl。配制方案fecd133为:按照0、20、40、60、80、100nm的浓度,tmz为0.1、0.2、0.3、0.4、0.5mm的浓度。加药48h后,弃去培养基,每孔加入100μl的10%tca,4℃固定1h,弃去固定液,使用蒸馏水洗三次,室温干燥;每孔加入100μl的0.4%srb,染色20min,平板震荡。弃去染液,1%乙酸洗三次,室温干燥。每孔加入100μl的10mm的trisbase,平板震荡10min溶解。酶标仪515nm的吸光度测量od值,如果od值大于2.0则增加trisbase进行稀释,重新测量。以0nm的三个孔od值的平均值作为对照,按照每孔的od值/对照的od值,其比例视为细胞活率,绘制细胞活率曲线,并使用graphprism软件计算每种细胞的ic50;
结果如图31所示,与对照细胞正常的胶质细胞(nha)相比,胶质瘤干细胞系gsc11、20171016b对fecd133更敏感;而作为对照的tmz在三种细胞中杀伤效果没有显著差异;
图32的结果显示,对结直肠癌(a图)和肝癌细胞(b图)通过流式筛选的cd133-(cd133阴性)和cd133+(cd133阳性)的细胞分别用fecd133处理,发现cd133+的细胞对fecd133更为敏感,而药物对正常结肠上皮细胞(hcoepic)和正常肝细胞(lo2)的杀伤效果较最弱。
实施例19:体内药敏实验
将1.0×106的gsc11的野生型细胞注射至5~6周龄的balb/c(nu/nu)裸鼠腹股沟大约两周左右,待肿瘤体积长到50mm3后,按照5mg/kggst、60mg/kgtmz、5mg/kgfecd133、50mg/kgfecd133和5mg/kg的fecd13联合60mg/kgtmz的量通过尾静脉注射至小鼠体内,共注射6次,实时监测接种部位的肿瘤的重量和体积,绘制生长曲线;六周后,待肿瘤体积长到一定比例,将裸鼠处死并解剖出肿瘤,进行肿瘤重量的称量和记录;
按实施例11颅脑原位注射成瘤实验2周后,按照5mg/kggst、60mg/kgtmz、5mg/kgfecd133和5mg/kg的fecd133联合60mg/kgtmz的量通过尾静脉注射至小鼠体内,共注射5次,待对照小鼠状态不佳,弥留之际,将裸鼠处死,取出脑组织,进行免疫组化染色,检测肿瘤生长情况;
结果如图37所示,用不同的药物处理之后,其裸鼠移植瘤大小如图a所示,50mgfecd133<tmz+fecd133<5mgfecd133<tmz<gst,b图是a图中肿瘤重量的统计结果,c图是a图中肿瘤体积的统计结果。
图38的结果显示用不同的药物处理之后,其原位注射成瘤能力如下:
tmz+fecd133<fecd133<tmz<gst;肿瘤的大小与小鼠的存活时间相反:tmz+fecd133>fecd133>tmz>gst。
序列表
<110>中国科学院昆明动物研究所
<120>人cd133蛋白1-108肽段的新用途
<160>20
<170>siposequencelisting1.0
<210>1
<211>108
<212>prt
<213>人(human)
<400>1
metalaleuvalleuglyserleuleuleuleuglyleucysglyasn
151015
serpheserglyglyglnproserserthraspalaprolysalatrp
202530
asntyrgluleuproalathrasntyrgluthrglnaspserhislys
354045
alaglyproileglyileleuphegluleuvalhisilepheleutyr
505560
valvalglnproargasppheprogluaspthrleuarglyspheleu
65707580
glnlysalatyrgluserlysileasptyrasplysprogluthrval
859095
ileleuglyleulysilevaltyrtyrglualagly
100105
<210>2
<211>21
<212>dna
<213>人工序列(artificial)
<400>2
atgtcattccgtaaagtaaac21
<210>3
<211>21
<212>dna
<213>人工序列(artificial)
<400>3
tcactttatgtttgagatctc21
<210>4
<211>21
<212>dna
<213>人工序列(artificial)
<400>4
gcactaccagagctaactcag21
<210>5
<211>21
<212>dna
<213>人工序列(artificial)
<400>5
agccagcacttgctcatttaa21
<210>6
<211>21
<212>dna
<213>人工序列(artificial)
<400>6
tctcaggaagtcctggaagat21
<210>7
<211>21
<212>dna
<213>人工序列(artificial)
<400>7
aagtgtgacgtggacatccgc21
<210>8
<211>22
<212>dna
<213>人工序列(artificial)
<400>8
ccggactcgtcatactcctgct22
<210>9
<211>21
<212>dna
<213>人工序列(artificial)
<400>9
acttgccaattctggttccca21
<210>10
<211>23
<212>dna
<213>人工序列(artificial)
<400>10
cggatgacaactttagtagaggc23
<210>11
<211>19
<212>dna
<213>人工序列(artificial)
<400>11
atggcaacagcgatcaagg19
<210>12
<211>22
<212>dna
<213>人工序列(artificial)
<400>12
gtactttgttggtgcaagctct22
<210>13
<211>20
<212>dna
<213>人工序列(artificial)
<400>13
agtcagcagagggacaggaa20
<210>14
<211>20
<212>dna
<213>人工序列(artificial)
<400>14
cttcgtacatggggagcact20
<210>15
<211>19
<212>dna
<213>人工序列(artificial)
<400>15
ggctcctggcaaaaggtca19
<210>16
<211>20
<212>dna
<213>人工序列(artificial)
<400>16
ctgcgtagttgtgctgatgt20
<210>17
<211>19
<212>dna
<213>人工序列(artificial)
<400>17
cacagatgcaaccgatgca19
<210>18
<211>18
<212>dna
<213>人工序列(artificial)
<400>18
ggtgccctgctgcgagta18
<210>19
<211>19
<212>dna
<213>人工序列(artificial)
<400>19
ctgccgctttgcaggtgta19
<210>20
<211>21
<212>dna
<213>人工序列(artificial)
<400>20
cattgtgggcaaggtgctatt21
- 还没有人留言评论。精彩留言会获得点赞!