一种富马酸丙酚替诺福韦片剂及其制备方法和有关物质的检测方法与流程
[0001]
本发明涉及药物制剂技术领域,尤其涉及一种富马酸丙酚替诺福韦片剂及其制备方法和有关物质的检测方法。
背景技术:
[0002]
乙型病毒性肝炎(乙肝)是由乙型肝炎病毒(hepatitis b virus,hbv)感染引起的,以乏力、食欲减退、恶心、呕吐、厌油、肝大及肝功能异常为主要临床症状,是一种发病率高、感染性强、严重危害人类健康的全身性感染病。目前,全球约有20亿人感染过乙型肝炎病毒,其中,3.5亿人成为慢性hbv携带者,全球每年有100万患者死于慢性乙型肝炎相关性疾病。乙肝已然成为严重威胁人类健康的难题,因此,研发一种治疗乙型肝炎病毒的药物至关重要。
[0003]
富马酸丙酚替诺福韦片剂是由美国吉利德科学公司研发,商品名velimdy,是一种乙型肝炎病毒(hbv)核苷酸逆转录酶抑制剂,于2016年11月获得fda批准上市,2018年11月在国内获批上市。目前国内只有进口的原研药品上市,尚无仿制品种获批。参考欧洲上市审评报告可知,原研制剂制备工艺为干法制粒,工艺繁琐且能耗较高,并且制备过程中会产生明显的物料粉碎粉尘,不利于环保。现有技术中,有研究人员曾尝试粉末直压的方法制备富马酸丙酚替诺福韦片,粉末直压的工艺产品硬度较大,产品溶出速度较慢,与原研一致性较差,且储存过程中杂质增长较大。因此,研发一种溶出速度快、与原研产品一致性好且稳定性好的富马酸丙酚替诺福韦片具有十分重要的意义。
技术实现要素:
[0004]
针对现有富马酸丙酚替诺福韦片存在的上述问题,本发明提供一种富马酸丙酚替诺福韦片剂及其制备方法和有关物质的检测方法。
[0005]
为解决上述技术问题,本发明提供的技术方案是:
[0006]
一种富马酸丙酚替诺福韦片剂,包括如下重量百分比的组分:富马酸丙酚替诺福韦12%-14%,交联剂10%-15%,稀释剂45%-58%,崩解剂5%-12%,硬脂富马酸钠1%-5%和磷酸氢钙5%-15%。
[0007]
富马酸丙酚替诺福韦原料的自身流动性差,原辅料不易混合均匀,本发明选择硬脂富马酸钠作为润滑剂,提高片剂生产中的可压性,从而有利于使制备的片剂圆整度高,粒径分布窄,硬度适中,并且对崩解时间和药物释放无影响,且硬脂富马酸钠与富马酸丙酚替诺福韦相容性好,可降低制剂在存储过程中有关物质的增加;磷酸氢钙作为填充剂,不仅可进一步改善物料的可压性,提高原料在片剂中的均匀度,同时协同硬脂富马酸钠、崩解剂和交联剂,还可提高片剂的溶出速率,改善崩解时限。本发明的富马酸丙酚替诺福韦片剂的配方,通过选用适量的硬脂富马酸钠作为润滑剂、适量的磷酸氢钙作为填充剂,并配合适量的交联剂、崩解剂和稀释剂,降低了富马酸丙酚替诺福韦原料在制备过程中的降解,压片质量
稳定,药品稳定性高,能更好发挥富马酸丙酚替诺福韦片剂的药效,提高其药物有效率,实现对原研品的有效替代。
[0008]
富马酸丙酚替诺福韦原料的粉体学测试结果如下表所示,所述原料为湖南明瑞制药有限公司生产的。
[0009]
批号堆密度振实密度卡尔系数(%)1703030.414g/cm30.571g/cm327.5
[0010]
由表中卡尔系数可知,富马酸丙酚替诺福韦原料的自身流动性较差。
[0011]
优选的,所述交联剂为羟丙甲纤维素。
[0012]
优选的,所述稀释剂为乳糖。
[0013]
选择乳糖和磷酸氢钙协同,能够改善物料的可压性,减少富马酸丙酚替诺福韦原料的剂量偏差,促进溶出和崩解。除此之外,还可提高制备的片剂的硬度和光洁度。
[0014]
优选的,所述崩解剂为羧甲基淀粉钠。
[0015]
选择羧甲基淀粉钠作为崩解剂配合羟丙基纤维素和硬脂富马酸钠,可提高富马酸丙酚替诺福韦片剂的崩解速度,提高溶出度,并达到与原研品溶出曲线一致。
[0016]
优选的,所述富马酸丙酚替诺福韦片剂还包括胃溶型包衣预混剂1%-6%。
[0017]
本发明所述胃溶型包衣预混剂为本领域常规的包衣预混剂,本领域技术人员可常规进行选择。
[0018]
本发明还提供一种富马酸丙酚替诺福韦片剂的制备方法,包括如下步骤:
[0019]
按照设计配比称取各组分,将各组分分别粉碎,过筛,然后将富马酸丙酚替诺福韦、65%-70wt%的交联剂、稀释剂、崩解剂、65%-70wt%的硬脂富马酸钠和磷酸氢钙混合均匀后,流化床制粒,制粒完成后加入剩余的交联剂和硬脂酰富马酸钙,混合均匀,得总混物料;根据中间体含量检测结果,将总混物料压片,包衣,得富马酸丙酚替诺福韦片剂。
[0020]
本发明提供的富马酸丙酚替诺福韦片剂的制备方法,采用流化床制粒法进行制粒,提高了制粒后富马酸丙酚替诺福韦的流动性,提高了可压性,从而提高了富马酸丙酚替诺福韦在产品中的均匀性,提高混合物的密度,减少单位重量的体积,并且制备的富马酸丙酚替诺福韦片剂具有适宜的硬度,溶出曲线与原研产品相似,同时,更重要的是有效地控制了富马酸丙酚替诺福韦的降解,保证药品的稳定性,与干法制粒相比,片剂的杂质稳定性也更加优异,药品稳定性高,可实现对原研品的替代。
[0021]
优选的,所述富马酸丙酚替诺福韦粉碎后过100目筛,其余辅料粉碎后过80目筛。
[0022]
优选的,流化床制粒工序中,流化床进风口的温度为20-40℃。
[0023]
优选的,压片工序中,布料盘转速为20-60hz,压片速度为1-3万片/小时,平均压力为2-10kn。
[0024]
优选的,包衣过程为:将所述胃溶型薄膜包衣剂配成质量浓度为8-12wt%的包衣液,在0.08-0.12mpa的压缩空气条件下搅拌15-20min,加入包衣机中包衣,增重为片芯总重量的1-6%;其中,包衣锅转速为3-10r/min,进风温度为60-70℃,出风温度为25-35℃,雾化压力为0.15-0.25mpa,喷浆速度为30-140rpm。
[0025]
本发明提供的富马酸丙酚替诺福韦片剂的制备方法,极大地提高了富马酸丙酚替诺福韦片的崩解速度和溶出速率,并有效提高了原料的分散均匀性,且制备的富马酸丙酚替诺福韦片剂的体外四种介质中溶出曲线均与原研一致,杂质含量低且稳定性好,提高了
临床应用的安全性。本发明提供的富马酸丙酚替诺福韦片剂的制备方法操作简单,可控性强,无明显的粉碎粉尘,对环境污染小,且工艺能耗低,易实现工业化大生产。
[0026]
本发明还提供了一种富马酸丙酚替诺福韦片剂中有关物质的检测方法,其特征在于,用高效液相色谱法进行检测,色谱条件为:
[0027]
色谱柱:十八烷基硅烷键合硅胶柱;
[0028]
uv检测器,检测波长260nm;
[0029]
流动相a:体积比为500:450:50的磷酸氢二钠缓冲液-四氢呋喃-乙腈,流动相b:体积比为950:40:10的磷酸氢二钠缓冲液-四氢呋喃-乙腈;
[0030]
洗脱方式为梯度洗脱,洗脱程序如下:
[0031]
0-1min,100%流动相a;
[0032]
1-5min,100%
→
90%流动相a,0%
→
10%流动相b;
[0033]
5-8min,90%
→
78%流动相a,10%
→
22%流动相b;
[0034]
8-65.01min,78%
→
100%流动相a,22%
→
0%流动相b;
[0035]
65.01-75min,100%流动相a。
[0036]
优选的,柱温为35℃,流速为1ml/min,进样体积为15μl。
[0037]
优选的,流动相a和流动相b中所述磷酸氢二钠缓冲液的浓度为0.02mol/l,ph为6.0。
[0038]
本发明提供的富马酸丙酚替诺福韦片剂中有关物质的检测方法,能够实现富马酸丙酚替诺福韦片剂中主成分与多个已知杂质、未知杂质的有效分离,准确定性及定量检测出富马酸丙酚替诺福韦片剂中多个已知杂质和未知杂质,且本发明提供的方法经方法学研究及验证,发现本发明的方法灵敏度、重现性较好,杂质的检测限和定量限低,能够实现对富马酸丙酚替诺福韦片剂中更多杂质的定性和定量检测,为提高和更好地控制索富马酸丙酚替诺福韦片剂的质量提供了可靠保障。
具体实施方式
[0039]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0040]
为了更好的说明本发明,下面通过实施例做进一步的举例说明。
[0041]
实施例1
[0042]
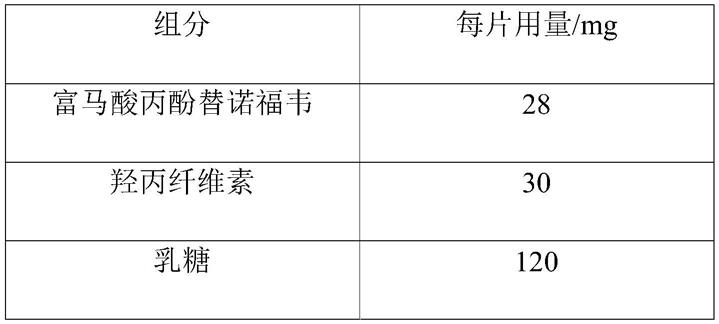
本实施例提供一种富马酸丙酚替诺福韦片剂,处方用量如下表所示:
[0043][0044][0045]
上述富马酸丙酚替诺福韦片剂的制备方法如下:
[0046]
将富马酸丙酚替诺福韦原料粉碎过100目筛备用,将其他辅料过80目筛备用。按照先辅料后主药的原则,按照处方量分别称取各组分,将称量好的富马酸丙酚替诺福韦、65wt%处方量羟丙纤维素、乳糖、羧甲基淀粉钠、磷酸氢钙以及68wt%处方量的硬脂富马酸钠混合均匀,流化床制粒,进风口的温度控制在20℃,制粒完成加入剩余的羟丙纤维素和硬脂富马酸钠,加入料斗混合机中,混合15min,混合转速10rpm。将混合好的物料检测中间体含量,根据含量确定片重。
[0047]
将中间体物料加入压片机的料斗中,压片。调试压片机压力与物料填充量,控制布料盘转速在40hz、冲头压力在10kn、车速在2万片/小时条件下下料试压片,片径8mm,片剂厚度2.8mm。压片机调试正常后开始压片,压好的片子经片子除尘器除尘后装入药用低密度聚乙烯袋密封。
[0048]
胃溶型薄膜包衣预混剂用水溶解,配成含固量10%的包衣液,在0.12mpa的压缩空气下搅拌15分钟。在包衣锅中包衣,增重6%。包衣参数:包衣锅转速3r/min,进风温度65℃,出风温度30℃,雾化压力0.2mpa,喷浆速度30rpm。
[0049]
实施例2
[0050]
本实施例提供一种富马酸丙酚替诺福韦片剂,处方用量如下表所示:
[0051]
[0052][0053]
上述富马酸丙酚替诺福韦片剂的制备方法如下:
[0054]
将富马酸丙酚替诺福韦原料粉碎过100目筛备用,将其他辅料过80目筛备用。按照先辅料后主药的原则,按照处方量分别称取各组分,将称量好的富马酸丙酚替诺福韦、70wt%处方量羟丙纤维素、乳糖、羧甲基淀粉钠、磷酸氢钙以及65wt%处方量的硬脂富马酸钠混合均匀,流化床制粒,进风口的温度控制在30℃,制粒完成加入剩余的羟丙纤维素和硬脂富马酸钠,加入料斗混合机中,混合15min,混合转速10rpm。将混合好的物料检测中间体含量,根据含量确定片重。
[0055]
将中间体物料加入压片机的料斗中,压片。调试压片机压力与物料填充量,控制布料盘转速在60hz、冲头压力在2kn、车速在3万片/小时条件下下料试压片,片径8mm,片剂厚度2.8mm。压片机调试正常后开始压片,压好的片子经片子除尘器除尘后装入药用低密度聚乙烯袋密封。
[0056]
胃溶型薄膜包衣预混剂用水溶解,配成含固量10%的包衣液,在0.1mpa的压缩空气下搅拌15分钟。在包衣锅中包衣,增重4%。包衣参数:包衣锅转速8r/min,进风温度65℃,出风温度30℃,雾化压力0.2mpa,喷浆速度80rpm。
[0057]
实施例3
[0058]
本实施例提供一种富马酸丙酚替诺福韦片剂,处方用量如下表所示:
[0059]
组分每片用量/mg富马酸丙酚替诺福韦28羟丙纤维素31乳糖106羧甲基淀粉钠15硬脂富马酸钠6磷酸氢钙11
胃溶型薄膜包衣预混剂10
[0060]
上述富马酸丙酚替诺福韦片剂的制备方法如下:
[0061]
将富马酸丙酚替诺福韦原料粉碎过100目筛备用,将其他辅料过80目筛备用。按照先辅料后主药的原则,按照处方量分别称取各组分,将称量好的富马酸丙酚替诺福韦、68wt%处方量羟丙纤维素、乳糖、羧甲基淀粉钠、磷酸氢钙以及70wt%处方量的硬脂富马酸钠混合均匀,流化床制粒,进风口的温度控制在40℃,制粒完成加入剩余的羟丙纤维素和硬脂富马酸钠,加入料斗混合机中,混合15min,混合转速10rpm。将混合好的物料检测中间体含量,根据含量确定片重。
[0062]
将中间体物料加入压片机的料斗中,压片。调试压片机压力与物料填充量,控制布料盘转速在20hz、冲头压力在7kn、车速在1万片/小时条件下下料试压片,片径8mm,片剂厚度2.8mm。压片机调试正常后开始压片,压好的片子经片子除尘器除尘后装入药用低密度聚乙烯袋密封。
[0063]
胃溶型薄膜包衣预混剂用水溶解,配成含固量10%的包衣液,在0.08mpa的压缩空气下搅拌15分钟。在包衣锅中包衣,增重1%。包衣参数:包衣锅转速10r/min,进风温度65℃,出风温度30℃,雾化压力0.2mpa,喷浆速度140rpm。
[0064]
对比例1
[0065]
将56.08g富马酸丙酚替诺福韦原料药和189.92g乳糖、120g微晶纤维素、28g交联羧甲基纤维素钠加入湿法混合制粒机中混合均匀,再加入6g硬脂酸镁作为润滑剂,混合均匀后直接压片,每片含富马酸丙酚替诺福韦25mg。将中间体物料加入压片机的料斗中,压片。压片参数:压片速度3000片/h,当前压片主压力0.08kn,片径8mm,片剂厚度2.8mm。胃溶型薄膜包衣预混剂用水溶解,配成含固量10%的包衣液。在包衣锅中包衣,增重3%。包衣参数:包衣锅转速8.0r/min,进风温度60℃,出风温度32℃,雾化压力0.1mpa,包衣液流速5ml/min。
[0066]
样品质量检测
[0067]
1.含量检测方法
[0068]
c18色谱柱,流动相:0.05mol/l磷酸氢二钠缓冲溶液(ph4.5)-乙腈(体积比50:50),检测波长260nm,柱温35℃,进样量20μl,流速1ml/min。
[0069]
2.有关物质检测方法
[0070]
c18色谱柱,流动相a:0.02mol/l磷酸氢二钠缓冲溶液(ph6.0)-四氢呋喃-乙腈(体积比500:450:50),流动相b:0.02mol/l磷酸氢二钠缓冲溶液(ph6.0)-四氢呋喃-乙腈(体积比950:40:10),检测波长260nm,柱温35℃,进样量20μl,流速1ml/min,梯度洗脱,洗脱顺序如表1所示。
[0071]
表1梯度洗脱顺序
[0072]
[0073][0074]
3.原料和制剂强制破坏试验结果
[0075]
1)热破坏供试品
[0076]
精密称取富马酸丙酚替诺福韦片研细粉末约218mg,置于50ml容量瓶中,60℃烘箱放置4h,加溶剂定容,摇匀,过滤,得制剂热破坏供试品溶液。
[0077]
精密称取空白辅料约190mg,置于50ml容量瓶中,60℃烘箱放置4h,加溶剂定容,得空白辅料热破坏供试品溶液。
[0078]
精密称取富马酸丙酚替诺福韦原料约28mg,置于50ml容量瓶中,60℃烘箱放置4h,加溶剂定容,摇匀,得原料热破坏供试品溶液。
[0079]
2)酸破坏供试品
[0080]
精密称取富马酸丙酚替诺福韦片研细粉末约218mg,置于50ml容量瓶中,加0.1n盐酸溶液1ml,放置10min,加0.1n氢氧化钠溶液1ml,加溶剂定容,摇匀,过滤,得制剂酸破坏供试品溶液。
[0081]
精密称取空白辅料约190mg,置于50ml容量瓶中,加0.1n盐酸溶液1ml,放置10min,加0.1n氢氧化钠溶液1ml,加溶剂定容,得空白辅料酸破坏供试品溶液。
[0082]
精密称取富马酸丙酚替诺福韦原料约28mg,置于50ml容量瓶中,加0.1n盐酸溶液1ml,放置10min,加0.1n氢氧化钠溶液1ml,加溶剂定容,得原料酸破坏供试品溶液。
[0083]
3)碱破坏供试品
[0084]
精密称取富马酸丙酚替诺福韦片研细粉末约218mg,置于50ml容量瓶中,加0.1n氢氧化钠溶液1ml,放置10min,加0.1n盐酸溶液1ml,加溶剂定容,摇匀,过滤,得制剂碱破坏供试品溶液。
[0085]
精密称取空白辅料约190mg,置于50ml容量瓶中,加0.1n氢氧化钠溶液1ml,放置10min,加0.1n盐酸溶液1ml,加溶剂定容,得空白辅料碱破坏供试品溶液。
[0086]
精密称取富马酸丙酚替诺福韦原料约28mg,置于50ml容量瓶中,加0.1n氢氧化钠溶液1ml,放置10min,加0.1n盐酸溶液1ml,加溶剂定容,得原料碱破坏供试品溶液。
[0087]
分别精密量取上述各供试品溶液,按照上述有关物质测定项下的方法测定,按外标法以峰面积计算强制破坏试验中的物料平衡情况,以未破坏样品作为对照品进行计算,结果如表2-表3所示。
[0088]
表2原料强制破坏试验结果
[0089][0090][0091]
表3制剂强制破坏试验结果
[0092][0093]
由上表可以看出,原料和制剂在酸性和碱性强制破坏条件下极不稳定,均被破坏产生较多杂质。
[0094]
4.溶出度检测方法
[0095]
为了选择合适的溶出介质,对富马酸丙酚替诺福韦在不同介质中的稳定性进行考察。
[0096]
溶出介质:ph1.2盐酸溶液、ph3.0邻苯二甲酸氢钾溶液、ph4.5醋酸盐缓冲液、ph6.8磷酸盐缓冲液、水。
[0097]
称取富马酸丙酚替诺福韦原料加对应的溶出介质,制成浓度为0.25mg/ml的溶液,在配置完成的第0、1、2、3、4、6、8、24小时,量取15μl,按照上述含量测定项下的方法测定,记录色谱图,结果如表4所示。
[0098]
表4
[0099]
峰面积水ph4.5ph6.8ph3.0ph1.20h139.8261137.0965139.5228139.209712.73661h140.2954138.6920139.5260139.54627.31572h139.6941136.8664137.2645138.44064.16173h139.8224136.8784137.0806136.72912.40064h139.8295136.9112136.9101136.84821.37366h137.7657136.7245136.4983136.0051——8h137.6555136.7017135.9770136.7548——24h136.5537132.0095135.2906135.6241——
均值138.9303136.4850137.2587137.3947——rsd1.001.411.121.07——
[0100]
由上表可以看出,富马酸丙酚替诺福韦在水、ph3.0、ph4.5、ph6.8介质中24h稳定,在ph1.2盐酸介质中降解严重,不稳定。因此,溶出曲线的测定选择ph3.0、ph4.5醋酸盐缓冲液和ph6.8磷酸盐缓冲液和水作为溶出介质。
[0101]
溶出度检测方法:取本品,照溶出度与释放度测定法(通则0931第二法)分别以ph3.0邻苯二甲酸氢钾溶液、ph4.5醋酸盐缓冲液、ph6.8磷酸盐缓冲液、水,各700ml为溶出介质,转速为每分钟75转,分别于5、10、15、20、30分钟取样检测。取溶液滤过,取续滤液作为供试品溶液;另取富马酸丙酚替诺福韦对照品适量,精密称定,加介质溶解并定量稀释制成每1ml中约含富马酸丙酚替诺福韦50μg的溶液,作为对照品溶液。按照上述含量测定项下的方法测定,计算每片的溶出量。
[0102]
影响因素试验:
[0103]
为考察本发明实施例制备的富马酸丙酚替诺福韦片的稳定性,按照中国药典2015年版四部通则《原料药与药物制剂稳定性试验指导原则》和ich q1a q1b的要求,对实施例1制备得到的产品与通过原研产品(gilead,made in canada,批号013162)分别进行了高温、高湿、光照的影响因素试验研究,试验条件如表5所示,试验结果如表6-表8所示。
[0104]
表5
[0105][0106]
表6影响因素高温试验结果
[0107]
[0108][0109]
表7影响因素高湿试验结果
[0110][0111][0112]
表8影响因素光照试验结果
[0113][0114]
由以上研究数据可知,本发明实施例制备的富马酸丙酚替诺福韦片杂质比原研品更低,且溶出度均能达85%以上,含量及溶出度均不低于原研产品。
[0115]
溶出曲线测定
[0116]
为考察本发明实施例制备的富马酸丙酚替诺福韦片的溶出度,参照溶出度与释放度测定法(中国药典2015年版四部通则0931第二法)要求,对实施例1、实施例2、实施例3及原研制剂(gilead,made in canada,批号013162)进行了加速及长期3月、6月溶出曲线的测定,长期及加速试验条件表9所示,试验结果如表10-表14所示。
[0117]
表9长期和加速实验条件
[0118]
加速试验放置于40℃
±
2℃、rh75%
±
5%长期试验放置于30℃
±
2℃、rh65%
±
5%
[0119]
表10 0月溶出曲线测定结果
[0120][0121]
表11长期3月溶出曲线结果:
[0122][0123]
表12加速3月溶出曲线测定结果
[0124]
[0125][0126]
表13长期6月溶出曲线测定结果
[0127][0128]
[0129]
表14加速6月溶出曲线测定结果
[0130][0131]
由以上数据可知,在4种溶出介质条件下,本发明采用流化床制粒法生产的富马酸丙酚替诺福韦片在10min溶出度均可达到85%以上,溶出曲线与原研品相似,能够实现与原研产品质量一致,能够实现对原研产品的较好替代。
[0132]
综上所述,本发明实施例制备的富马酸丙酚替诺福韦片具有合适的硬度、重量差异小、片面光洁、溶出度曲线与原研产品溶出曲线相似,质量稳定性好,能够实现对原研产品的替代;制剂生产工艺简单,无明显的粉碎粉尘,对环境污染小,且工艺能耗低,易实现工业化大生产。
[0133]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换或改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1