一种基于宏条形码的家畜采食组成校正模型的构建方法与流程
本发明属于分子生物学领域和畜牧学领域,具体地,本申请提供了一种基于宏条形码的家畜采食组成校正模型的构建方法
背景技术:
放牧家畜的生产性能和营养状况与牧草摄入量密切相关。在放牧条件下,准确掌握家畜的采食成分与采食量极其关键,能够为明晰家畜营养状况和预测家畜生产性能,进而为制定草地管理决策、优化牧草资源配置等提供有效的数据支撑和理论指导。天然草地上的牧草资源丰富,种类繁多,食草家畜摄入的植物种类非常多元化,而且会对特定的牧草种类存在一定的偏好选择,准确测定食草家畜的采食组分仍然面临诸多困难与挑战。
国内外常用于估测食草动物采食成分的方法众多,主要有模拟采食法、牧草采食法、食道瘘管法、瘤胃内容物法、粪便显微分析法、植物蜡层指示剂法和近红外光谱法。饱和烷烃技术目前被认为是最准确、最客观的方法,它是将饱和烷烃作为内源指示剂,通过测定家畜所采食各牧草的链烷种类、含量,与粪便中相应链烷的种类、含量建立回归方程,从而估算动物对各牧草的采食比例。该方法能够准确地估测日粮组分简单(4~8种)的动物采食组分。但是当牧草中的烷烃浓度较低或者链烷模式相似时,随着家畜食物组成复杂化,其估测准确性大大降低。此外,蜡层指示剂的特征模式及其回收率也会因季节和地域等因素有所差异,直接影响季节性采食估测结果的准确性。
dna条形码技术(dnabarcoding),是指用基因组内一段标准的、相对较短的dna片段来鉴定物种或者其变异类型的一项分子鉴定新技术。该技术摆脱了传统鉴定方法依赖长期经验的障碍,通过建立标准数据库,可实现对物种的快速准确鉴定,是分子鉴定方法学上的创新。然而dna条形码技术依赖传统的sanger测序技术,尚无法实现地对多个体多物种的混合样品进行高效快速分类及评估,而且从陈旧材料难以获得完整、高质量的dna模板。基于高通量测序技术(也称二代测序技术)的dna宏条形码技术有望克服这一难题。
dna宏条形码技术,也叫dna复合条形码技术,是指利用高通量技术同时获得很多物种的条形码基因扩增子序列,借助生物信息学分析手段对群落的分类单元组成进行鉴定。它的工作原理是:(1)从环境样本中提取dna;(2)选取合适的dna条形码作为分子标记;(3)利用条形码通用引物从环境样本中获得扩增子;(4)利用合适的高通量测序平台,对大量的扩增子进行高通量测序;(5)将测序得到的序列与参考数据库进行比对,得到物种注释信息;(6)对测序结果进行生物信息学分析。
近些年来,dna宏条形码技术(dnametabarcoding)在量化膳食成分上的研究取得很大进展。研究表明,dna宏条形码技术因其快速、操作简单、健壮等优势,适合测定食草类动物的食物组分。但是基于植物dna条形码分析食草动物的饮食面临的困难更大。在动物食物组分的鉴定研究中,人们关注的是宏条形码技术是否能够针对动物所消耗的成分做出足够精确的估计。我们前期通过绵羊舍饲试验评估了dna宏条形码技术在估测放牧家畜采食组分上的准确性。发现,通过从粪便中扩增植物its2条形码并进行illuminamiseq测序,能够定性鉴定绵羊所采食的牧草种类。并且初步证实绵羊粪便中回收得到的牧草dna序列比例与绵羊实际消耗的牧草比例存在线性关系。
而由于食草动物消耗的种类及其消化率也存在差异等复杂因素,测定结果存在偏差在所难免。先前已经针对绵羊消耗的部分牧草比例构建的定量预测模型其准确性仍需要进一步补充和完善。
技术实现要素:
一方面,本发明提供了一种基于宏条形码的家畜采食组成校正模型的构建方法:包括:(1)选取源自不同牧草制备模拟日粮,以此作为校正样品,提取混合样本dna、its2条形码pcr扩增、扩增子illuminamiseq双末端测序,获得校正样品的原始测序数据;
(2)对原始测序数据进行过滤和质控处理,将有效序列进行操作分类单元otu聚类;
(3)将otu代表序列与ncbi数据库blastn比对,在属或种水平上完成物种注释;
(4)使用宏条形码技术对每个混合样本的牧草基因序列进行测定,得到牧草dna序列的相对比例测定值,分析获得模拟日粮的组分;
(5)利用线性回归方法对模拟日粮各组分的测定值、各组分的干物质比例真实值进行关联,建立日粮组成的校正模型。
进一步地,提取混合样本dna包括液氮研磨步骤。
进一步地,its2条形码pcr扩增时所用引物为rd5-its2:tcctccgcttattgatatgc以及rb1-its2f:cgatacttggtgtgaattgcag。
进一步地,步骤(2)中的质控处理包括(i)过滤read尾部质量值20以下的碱基,以50bp为一个滑动窗口,若窗口内序列的平均质量值低于20,从窗口开始截去后端碱基。过滤质控后读长小于50bp的reads;(ii)根据pereads之间的重叠关系,将成对reads拼接成一条序列,相互拼接上的序列之间,重叠区最小重叠长度为10bp,拼接序列的重叠区允许的最大错配比率为0.2,筛选不符合序列;(iii)根据序列首尾两端的barcode和引物区分样品,并调整序列方向,barcode允许的错配数为0,最大引物错配数为2,去掉无法拼接的序列,最后去除barcode标签及引物序列。
进一步地,步骤(2)中的otu聚类使用usearchversion软件,按照97%相似性进行,在聚类过程中使用uchime软件识别并去除嵌合体序列。
进一步地,还包括回归校正步骤。
进一步地,回归校正中使用kulczyński相似系数(ksi,%)来评价准确性,其计算公式如下:
其中,ci是模拟日粮中第i个牧草的真实值与估测值之间的较小值;(ai+bi)是每一种牧草组分的真实值与估测值的比例之和。
另一方面,本申请提供了上述方法在家畜采食组成测定中的应用。
进一步地,所述家畜为绵羊。
本申请中的牧草可根据具体需要和条件选择各种已知或未知的种类,包括但不限于碱茅属(puccinellia)、狗牙根属(cynodon)、早熟禾属(poa)、黑麦草属(lolium)、雀麦属(bromus)、赖草属(leymussp.)、苜蓿属(medicago)、车轴草属(trifolium)、菊苣属(cichorium)9个属等属的,碱茅(puccinelliadistans)、狗牙根(cynodondactylon)、草地早熟禾(poapratensis)、黑麦草(loliumperenne)、直立雀麦(bromuserectus)、紫花苜蓿(medicagosativa)、红三叶(trifoliumpratense)、菊苣(cichoriumintybus)、狐尾三叶草(trifoliumrubens)、鳞状三叶草(trifoliumsquamosum)大麦状雀麦(bromushordeaceus)等具体种。
本申请中的家畜包括各种食草或杂食类家畜,包括但不限于羊如山羊、绵羊或肉用羊、毛用羊、肉毛兼用羊;牛如奶牛、肉牛;猪;马;驴等。
本申请中的分子生物学操作方法,如dna提取、pcr方法等本领域技术人员可以根据本领域常规技术手段(包括但不限于《分子克隆》,萨姆布鲁克等工具书)选择和实施。并可以根据需要选用各种成品试剂盒。
本发明的有益效果包括:本发明建立了模拟日粮牧草组分的回归校正模型,包括总体回归方程和单个回归方程。与模拟日粮组分的实际比例进行对比,利用相似系数ksi对估测值和校正值进行准确性分析,发现用总体回归方程和单个回归方程校正之后,采食组分的测定结果准确性显著提高。用构建的模型对舍饲绵羊的采食组分进行校正,校正之后测定结果的准确性有了明显提高。
附图说明
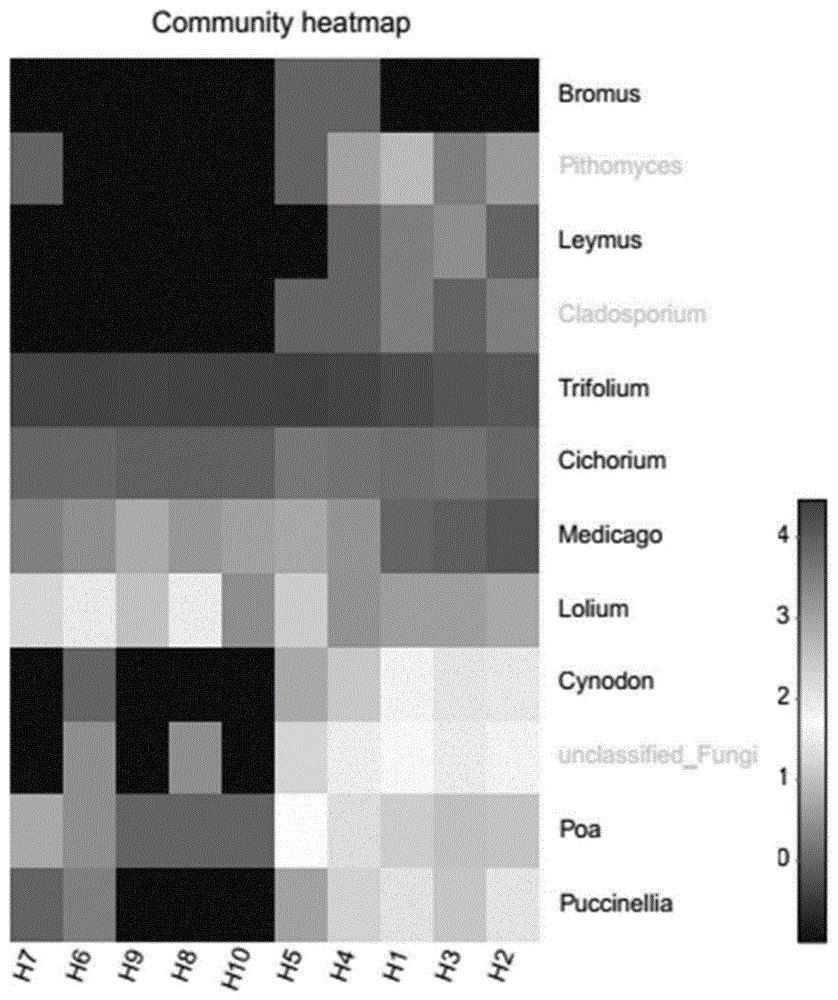
图1为模拟日粮中牧草的相对丰度(属水平);
图2为模拟日粮中牧草的相对丰度(种水平);
图3为模拟日粮中牧草组分的估测值与真实值之间的回归关系;
图4为模拟日粮中各个牧草种类的线性回归模型;
图5为模拟日粮处理中基于宏条形码技术的牧草组分估测值与真实值间的相似系数(ksi)牧草组分的估测值包括未校正(灰色方形)、总体回归方程(ce-1,红色圆形)和单个回归方程(ce-2,蓝色三角)。d:模拟日粮;rc:回归校正;d×r:模拟日粮和回归校正方法的交互作用。星号***表示在p<0.001水平上有显著差异;
图6为日粮处理1、2、3和4中基于dna条形码技术的采食成分估测值与测量值间的相似系数(ksi)。粪便中检测的采食组分的估测值包括未校正(灰色圆圈)、校正方程1(黑色方形)和校正方程2(黑色三角形)。d:日粮;rc:回归校正;d×r:日粮和回归校正方法的交互作用。星号***表示在p<0.001水平上有显著差异;
图7为日粮处理1、2、3和4中基于宏条形码和饱和烷烃两种方法的采食成分估测值与真实值间的相似系数(ksi)比较。相似系数计算所基于的校正方法包括未校正(灰色圆圈)、校正方程1(黑色方形)、校正方程2(蓝色三角形)、个体回收率(黑色圆圈)、处理回收率(红色圆圈)和总体回收率(黑色三角形)。d:日粮;cm:校正方法;d×cm:日粮和校正方法的交互作用。星号***表示在p<0.001水平上有显著差异。
具体实施方式
实施例1对模拟日粮中牧草组分的测定
将收集的9种牧草经液氮充分研磨后,按照设定比例(干物质比例)充分混匀,作为模拟日粮,比例见下表1。其中包括10个处理,每个处理重复4次,共40个牧草混合样本。每个混合牧草样本约0.5g装入到离心管内,置于液氮罐中保存。
表1模拟日粮的组成
经液氮磨碎混匀后的模拟日粮样品用植物基因组dna提取试剂盒(dnasecureplantkit)提取样品植物总dna。以上述dna为模板,用文献中已报道的引物rd5-its2和rb1-its2f(表2)pcr扩增植物its2序列,且对于每个样本分别加上8bp的标签序列,用于区分测序分析时各个数据的样本来源。
表2illuminamiseq测序引物
pcr扩增反应在abi
使用axyprepdna凝胶回收试剂盒(axygenbioscience,unioncity,ca,u.s.)从2%琼脂糖凝胶中回收pcr产物,用quantifluortm-st蓝色荧光定量系统(promega,u.s.)进行检测定量。之后将纯化后的pcr扩增子等比例混合。然后根据illuminamiseq测序平台的标准流程进行双端(2×300bp)测序。
用qiimeversion1.17软件对原始fastq文件进行严格的过滤和质控处理。数据过滤方法和参数如下:(i)过滤read尾部质量值20以下的碱基,以50bp为一个滑动窗口,若窗口内序列的平均质量值低于20,从窗口开始截去后端碱基。过滤质控后读长小于50bp的序列(reads);(ii)根据pereads之间的overlap关系,将成对reads拼接成一条序列,相互拼接上的序列之间,重叠区最小overlap长度为10bp。拼接序列的overlap区允许的最大错配比率为0.2,筛选不符合序列;(iii)根据序列首尾两端的barcode和引物区分样品,并调整序列方向,barcode允许的错配数为0,最大引物错配数为2。去掉无法拼接的序列,最后去除barcode标签及引物序列。
原始序列文件经过滤和质控处理后,形成一个新的序列文件,且每条序列都有一个特定的标签。使用usearchversion7.1软件,按照97%相似性对序列进行操作分类单元(operationaltaxonomicunits,otu)聚类,在聚类过程中使用uchime软件识别并去除嵌合体序列,得到otu的代表序列。将所有优化序列匹配至otu代表序列,选出与otu代表序列相似性在97%以上的序列,生成otu表格。
分类学比对时,用genbank“nucleotidecollection(nr/nt)”数据库,利用在线blastn工具,按照序列同源性最高原则,对每一个otu进行物种注释。若两条或两条以上代表性otu序列所得分数相同,或者没有比对到种水平上,则在更高水平上(属或科水平)对该序列进行物种分类学注释。
将otu表中的不相关序列(真菌)进行剔除,然后进行模拟日粮组成分析。otu序列数大于等于2的,认定为有效序列。序列数小于2的,可能是由于its2测序错误所导致,后续分析不予考虑。对dna序列数进行对数转换后,计算得到模拟日粮样本中每种牧草dna序列的相对比例,进而分析模拟日粮的组分。
对40份混合牧草样品(模拟日粮)中的its2条形码进行测序后,共得到1,473,296优质序列,通过聚类并去除真菌类后,得到40个otus。对全部样品中的有效序列进行归类操作分析,在属和种的水平统计所在分类单元对应的牧草种类及相对丰度(图1和图2)。对otu代表序列进行注释,结果表明样品中共有碱茅属(puccinellia)、狗牙根属(cynodon)、早熟禾属(poa)、黑麦草属(lolium)、雀麦属(bromus)、赖草属(leymussp.)、苜蓿属(medicago)、车轴草属(trifolium)、菊苣属(cichorium)9个属。其中丰度最高的3个分别是车轴草属、菊苣属、和苜蓿属,占总otus的62.5%。其中有36个otus鉴定到种的水平,分别是碱茅(puccinelliadistans)、狗牙根(cynodondactylon)、草地早熟禾(poapratensis)、黑麦草(loliumperenne)、直立雀麦(bromuserectus)、紫花苜蓿(medicagosativa)、红三叶(trifoliumpratense)、菊苣(cichoriumintybus)、狐尾三叶草(trifoliumrubens)、鳞状三叶草(trifoliumsquamosum)大麦状雀麦(bromushordeaceus)11个植物种。
实施例2回归模型构建和模拟日粮组分的准确性检验
采用线性回归分析确定通过测序获得的dna序列其相对比例与模拟日粮中牧草比例二者之间的定量关系,得到的回归方程用于校正直接估测结果。
对高通量测序得到的采食组分的直接估测结果进行回归校正,估测和校正后结果的准确性采用kulczyński相似系数(ksi,%)来评价。其计算公式如下:
其中,ci是模拟日粮中第i个牧草的真实值与估测值之间的较小值;(ai+bi)是每一种牧草组分的真实值与估测值的比例之和。
应用illuminamiseq测序后得到每个otu的序列数目,经过对数转化后计算各个牧草在不同混合牧草样品中的相对比例,然后与混合牧草样品中的真实比例进行比较。然后对九种牧草dna序列的相对比例与它们在模拟日粮中的实际比例进行回归分析,结果表明,二者之间存在极显著的线性回归关系(图3)。采用spss软件对参数进行拟合,得到模拟日粮中牧草组分的总体回归预测方程y=0.425x+0.064。
共设置10种不同比例的模拟日粮,其中包含9种常见草种。针对这9种牧草种类分别建立了单个的线性回归方程并进行方差分析,如图4所示。这些方程分别是:碱茅(pd),y=0.946x+0.098;狗牙根(cd),y=0.999x+0.088;早熟禾(pp),y=0.617x+0.044;黑麦草(lp),y=0.377x+0.022;雀麦(be),y=0.437x+0.038;羊草(lc),y=0.650x+0.059;紫花苜蓿(ms),y=1.727x-0.189;红三叶(tp),y=1.018x-0.065;菊苣(ci),y=0.055x+0.095。
通过dna宏条形码技术估测获得日粮中各种牧草的相对比例,利用总体回归方程(ce-1)和单个回归方程(ce-2)这两种模型,对估测的结果进行回归校正。表1展示了模拟日粮中9种牧草的实际测定值(oberserved)、估测值(estimated)、回归校正值(ce-1和ce-2)。采用相似系数ksi对估测结果以及两种方法校正后的结果的准确性进行评估。图5是三种结果的相似系数ksi的方差分析结果。可以看出,日粮处理(d)、校正方法(rc)以及二者之间的交互作用(d×rc)对ksi有极显著影响(p<0.001)。
显然,经单个回归方程和总体回归方程校正后,对日粮组分估测结果的准确性明显提高。直接估测结果(未经校正的)的ksi值为66.02%~76.18%,平均值为70.15%;采用总体回归方程(ce-1)校正的ksi值为73.84%~86.37%,平均值为80.42%;经单个回归方程(ce-2)校正的ksi值为85.84%~93.29%,平均值为90.58%。
实施例3估测并校正舍饲绵羊日粮组分
利用本发明中its2条形码测序的方法对绵羊采食组分进行了估测。同样采用ksi对该方法的准确性进行了方差检验(图6)。方差分析结果表明,日粮处理、回归方程校正方法以及二者之间的交互作用对ksi有着极显著作用(p<0.001)。经过两种回归方程校正之后,ksi明显增加(p<0.001)。三种估测结果相比较而言,可以发现采用第二种校正方程(ce-2)校正后,ksi最高(88.41%),其次是第二种校正方程ce-1(70.24%),未经校正的ksi值最低(57.97%)。
从日粮组分的角度分析,直接利用dna宏条形码技术估测得到的采食组分,估测值与真实值之间的相似性较低(46.98%~67.99%),且随着采食组分的增加ksi明显降低。而经ce-1(66.89%~72.03%)和ce-2(84.5%~92.0%)校正后,随着采食组分的增加,ksi比较平稳,并没有呈现骤然下降的趋势。
实施例4与饱和烷烃技术的应用效果对比
本研究分别用宏条形码和饱和烷烃技术对绵羊的采食组分进行了估测,并利用相似系数ksi对这两种技术的准确性进行了评估。图7呈现了在四种日粮处理下,采用6种方法对绵羊采食组分的估测值与真实值之间的相似性。结果显示,6种估测结果的准确性顺序为:个体回收率(i)>处理回收率(t)>校正方程2(ce-2)>总体回收率(g)>校正方程1(ce-1)>直接估测值(estimated)。表2列出了这6种校正方法的多重比较结果。可以看出,相似系数ksi在校正方法2和处理回收率二者之间并没有显著差异。
粪便中指示剂的回收率差异从单只绵羊回收率、处理回收率到总体回收率呈递增趋势变化,而且回收率校正之后提高了采食成分估测结果的准确性。然而,由于日粮组分显著影响粪便中长链醇的回收率,所以由于日粮组分引起的长链醇的总体回收率很大程度地偏离了处理平均回收率。所以总体回收率仅对其中的一种日粮组分进行了准确估测。根据单只绵羊回收率校正后的估测结果最为准确,然而在野外环境下难以操作]。与单只绵羊回收率校正的方法相较而言,虽然同一日粮处理回收率校正的估测结果准确性有所下降,但是影响很小。因此建议使用同日粮处理回收率校正法。
同样,采用相似系数ksi对dna条形码技术对绵羊采食组分的估测结果进行了评价。首先我们利用两个校正方程对估测结果加以校正。经过回归方程校正之后发现,与校正前相比而言,ksi值呈明显增加,即估测结果的准确性明显提高(图7)。而且,与饱和烷烃方法的结果有所不同,就是随着采食组分的增加,ksi的变化幅度比较小,受采食组分的影响较小。由此可以预见,对于复杂组分的估测而言,宏条形码技术比饱和烷烃的准确性更高。
在饱和烷烃技术的研究结果中,从实际应用角度出发,我们推荐使用处理回收率对烷烃的回收率进行校正。而图7和表2中的方差分析结果表明,宏条形码技术估测的结果经第二种方法校正之后,二者的准确性并没有显著差异,这也证实了本发明在估测绵羊采食组分上的应用效果和潜能。
表4于六种校正方法的估测值与真实值之间的相似系数
此实施例仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应该以权利要求的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!