一种融合多源生物信息的关键蛋白质识别方法
1.本发明涉及一种融合多源生物信息的关键蛋白质识别方法,主要是在蛋白质相互作用网络中融合蛋白质的多个生物信息的关键蛋白质识别技术,特别涉及蛋白质相互作用网络中融合了网络拓扑信息和蛋白质生物属性识别蛋白质复合物的方法,属于生物信息技术领域。
背景技术:
2.已有研究成果表明,人类疾病的发生和发展都与蛋白质的动态变化密切相关。例如,乳腺癌和肠癌的基因组是由少数常见突变基因和大量的频率较低的突变基因组成。不同蛋白质对生命活动的重要性是不一样的。winzeler[a]在《science》发表文章给出了关键蛋白质的定义,他认为关键蛋白质是指通过基因剔除式突变将其移除后造成有关蛋白质复合物功能丧失,并导致生物体无法生存或发育的蛋白质。关键蛋白质的识别能够从系统水平上为生物学、医学等提供有价值的信息。
[0003]
在本发明提出之前,关键蛋白质识别领域,最开始是通过一系列基于网络拓扑结构特征的关键蛋白质识别方法,例如,度中心性(dc)、接近度中心性(cc)、介数中心性(bc)、子图中心性(sc)、局部平均联通度(lac)等方法,但是这些方法识别关键蛋白质的缺点是:(1)只考虑了网络本身所具有的拓扑特征,而忽略了蛋白质所固有的生物属性特征。(2)通过生物实验所获得的ppi网络存在噪声,使得蛋白质相互作用数据存在假阳性。
技术实现要素:
[0004]
本发明的目的就在于克服上述缺陷,提供一种融合多源生物信息的关键蛋白质识别方法,该识别关键蛋白质的方法,是先构建动态ppi子网络,随后在ppi子网络中对于任意一节点计算出它与所有邻居节点之间的点聚类系数值之和、基因共表达值之和、go注释相似度值之和、以及细胞核位置得分值之和。接着将上述的值相加,作为蛋白质节点的关键性值,最后按照关键性值降序排序输入。
[0005]
本发明是这样实现的:一种融合多源生物信息的关键蛋白质识别方法,其主要技术特征在于如下步骤:
[0006]
(1)输入ppi网络和基因表达数据、go(蛋白质生物功能的注释属性)注释数据、亚细胞定位数据;
[0007]
(2)根据静态ppi网络和蛋白质基因表达值数据集,利用3σ法则构建多个动态子网络;
[0008]
(3)对于任一子网中的任一节点u来说,计算出该节点的点边缘聚类系数值decc(u,v),最后求该节点与其所有邻居之间的点聚类系数值之和sum_decc(u);
[0009]
(4)对于任一子网中的任一节点u来说,计算出该节点与邻居节点之间的基因共表达值pcc(u,v),最后求该节点与其所有邻居之间的共表达系数之和sum_pcc(u);
[0010]
(5)对于任一子网中的任一节点u来说,计算出该节点的亚细胞定位得分值sls
(u),最后求该节点与其邻居之间的亚细胞定位得分值之和sum_sls(u);
[0011]
(6)对于任一子网中的任一节点u来说,计算出该节点与邻居节点之间的go注释值go(u,v),最后求该节点与其所有邻居之间的共表达系数之和sum_go(u);
[0012]
(7)对于每一个节点u来说,将上述的属性值进行相加,得到蛋白质u的最终关键性得分值ess_pro(u);
[0013]
(8)最后将蛋白质节点按照ess_pro(u)的值从大到小排序输出。
[0014]
进一步,所述步骤(2)根据静态ppi网络和蛋白质基因表达值数据集,利用3σ法则构建多个动态子网络;根据基因表达值数据集,可以计算出每个蛋白质的活跃阈值tg,再结合静态网络的拓扑属性,可以将静态网络根据时间点划分为多个动态子网络,根据基因表达值数据集,可以计算出每个蛋白质的活跃阈值tg,再结合静态网络的拓扑属性,可以将静态网络根据时间点划分为多个动态子网络;每个蛋白质的活跃阈值tg计算过程如公式(1)-(4)所示:
[0015][0016][0017][0018]
tg=ug+3ρg(1-fg)
ꢀꢀꢀ
(4)
[0019]
蛋白质的基因表达值随时间变化而变化,在公式(1)中,n表示每个蛋白质基因的表达值的数量,gi表示在i时刻某个蛋白质基因表达值,ug表示某个蛋白质基因表达值的平均值,在公式(2)中,ρg表示的是某个蛋白质基因表达值的标准差的值,在公式(3)中,fg表示某个蛋白质一组基因表达值的波动性;最后在公式(4)中,tg表示是某个蛋白质的活跃阈值,也就是说,如果在某个时刻,蛋白质的基因表达值大于这个阈值tg,就可以说在该时刻,蛋白质是活跃的、表达的。
[0020]
进一步,所述步骤(3)对于任一子网中的任一节点u来说,计算出该节点的点边缘聚类系数值decc(u,v),最后求该节点与其所有邻居之间的点聚类系数值之和sum_decc(u);其中decc(u,v)、sum_decc(u)的计算公如下(5)、(6)表示:
[0021][0022][0023]
[0024][0025]
其中在公式(5)中,cn
u,v
表示结点u,v共同邻居的个数,ku,kv分别表示结点u,v的度,dccu,dccv表示结点u,v的点聚集系数,其计算如上述公式(7)所示,在公式(7)中kv表示结点v的度,nv表示由结点v的邻居结点之间组成的边数目;最后借助公式(8)对节点u和所有邻居节点v之间decc(u,v)值求和,其中v∈nu表示与u相连的所有邻居节点的集合。
[0026]
进一步,所述步骤(4)对于任一子网中的任一节点u来说,计算出该节点与邻居节点之间的基因共表达值pcc(u,v),最后求该节点与其所有邻居之间的共表达系数之和sum_pcc(u);其中两个蛋白质节点之间的共表达系数pcc(u,v)值计算如公式(8)所示,sum_pcc(u)计算如公式(9)所示:
[0027][0028]
在公式(8)中,u={u1,u2...un},v={v1,v2...vn}分别表示的是蛋白质u和蛋白质v的n个基因表达值;u’和v’分别表示其基因表达值的平均值;如果两个相互作用的蛋白质u,v基因的共表达程度越高,计算出来的pcc(u,v)值也就越大;在公式(9)中,v∈nu表示与u相连的所有邻居节点的集合。
[0029]
进一步,根据权利要求1所述的融合多源生物信息的关键蛋白质识别方法,其特征在于:所述步骤(5)对于任一子网中的任一节点u来说,计算出该节点和邻居之间的亚细胞定位得分值sls(u,v),最后求该节点与其邻居之间的亚细胞定位得分值之和sum_sls(u);其中蛋白质节点的亚细胞定位得分sls(u)值计算如公式(10)所示,sum_sls(u)计算如公式(11)所示:
[0030][0031][0032]
其中在公式(10)中|n|、|m|表示节点u、v在细胞核中出现的次数,在公式(11)中v∈nu表示与u相连的所有邻居节点的集合。
[0033]
进一步,所述步骤(6)对于任一子网中的任一节点u来说,计算出该节点与邻居节点之间的go注释值go_sim(u,v),最后求该节点与其所有邻居之间的共表达系数之和sum_go(u);其中蛋白质节点之间的go注释相似度值go_sim(u,v)计算如公式(12)所示,sum_go(u)计算如公式(13)所示:
[0034][0035]
[0036]
其中在公式(12)中,分子中的绝对值表示蛋白质u和v具有相同的go注释的数量;分母|gou|和|gov|表示蛋白质u和v拥有go注释的数量;在公式(13)中,v∈nu表示与u相连的所有邻居节点的集合。
[0037]
进一步,所述步骤(7)对于每一个节点u来说,将上述的属性值进行相加,得到蛋白质u的最终关键性得分值ess_pro(u),最后将ess_pro(u)的值从大到小排序;其中蛋白质节点的关键性值如下公式(14)所示:
[0038]
ess_pro(u)=sum_pcc(u)+sum_go(u)+sum_sls(u)+sum_decc(u)
ꢀꢀꢀ
(14)
[0039]
其中在公式(14)中的sum_pcc(u)、sum_go(u)、sum_sls(u)、sum_decc(u)分别是上述步(3)-(6)中所求的值。
[0040]
进一步,所述步骤(8)最后将蛋白质节点按ess_pro(u)的值从大到小排序输出,就是将步骤7中所求的蛋白质关键性值进行降序排序,然后从大到小将蛋白质节点输出,这就是最终的实验结果。
[0041]
本发明的优点和效果在于:该方法不但考虑了蛋白质相互作用网络的拓扑特征,同时也更多地考虑了蛋白质的生物属性,进而克服数据噪声高所带来的负面影响。融合多个生物属性提高了识别关键蛋白质的准确性,同时使预测结果更加准确,提高了预测的效率。扩展了该技术在生物信息领域的应用范围和实用性。
附图说明
[0042]
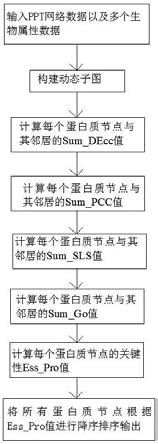
图1是本发明一种融合多源生物信息的关键蛋白质识别方法的流程示意图。
[0043]
图2是本发明在dip数据集上前1%关键蛋白质数量的对比图。
[0044]
图3是本发明在dip数据集上前5%关键蛋白质数量的对比图。
[0045]
图4是本发明在dip数据集上前10%关键蛋白质数量的对比图。
[0046]
图5是本发明在dip数据集上前15%关键蛋白质数量的对比图。
[0047]
图6是本发明在dip数据集上前20%关键蛋白质数量的对比图。
[0048]
图7是本发明在dip数据集上前25%关键蛋白质数量的对比图。
[0049]
图8是本发明在mips数据集上前1%关键蛋白质数量的对比图。
[0050]
图9是本发明在mips数据集上前5%关键蛋白质数量的对比图。
[0051]
图10是本发明在mips数据集上前10%关键蛋白质数量的对比图。
[0052]
图11是本发明在mips数据集上前15%关键蛋白质数量的对比图。
[0053]
图12是本发明在mips数据集上前20%关键蛋白质数量的对比图。
[0054]
图13是本发明在mips数据集上前25%关键蛋白质数量的对比图。
[0055]
图14是本发明在krogan数据集上前1%关键蛋白质数量的对比图。
[0056]
图15是本发明在krogan数据集上前5%关键蛋白质数量的对比图。
[0057]
图16是本发明在krogan数据集上前10%关键蛋白质数量的对比图。
[0058]
图17是本发明在krogan数据集上前15%关键蛋白质数量的对比图。
[0059]
图18是本发明在krogan数据集上前20%关键蛋白质数量的对比图。
[0060]
图19是本发明在krogan数据集上前25%关键蛋白质数量的对比图。
具体实施方式
[0061]
下面结合附图和具体实施方式对本发明进行详细说明。
[0062]
本发明的技术思路是:将蛋白质多个生物属性和蛋白质相互作用网络的拓扑特性相结合,首先利用3σ法则和蛋白质基因表达值数据集,将静态ppi网络转化为多个动态ppi子网络,然后在ppi子网络中对于任意一节点计算出它与所有邻居节点之间的点聚类系数值之和、基因共表达值之和、go注释相似度值之和、以及细胞核位置得分值之和。接着对每一个蛋白质节点将上述的值相加,作为该蛋白质节点的关键性值,最后按照关键性值降序排序输入。融合多个生物属性和拓扑特性有助于理解未知关键蛋白质的功能,对于解释特定功能的分子机制有着重要意义,同时能够对药物靶细胞设计等提供重要的理论依据。所以融合多源生物信息的关键蛋白质识别方法很自然地适用于关键蛋白质的预测。
[0063]
如图1所示,一种融合多源生物信息的关键蛋白质识别方法,包括以下步骤:
[0064]
步骤1:输入ppi网络和基因表达数据等生物属性信息数据、亚细胞定位数据。
[0065]
步骤2:根据静态ppi网络和蛋白质基因表达值数据集,利用3σ法则构建多个动态子网络。
[0066]
基因表达谱数据集:在蛋白质基因表达谱数据集中,每个蛋白质有36个活动点,即每个蛋白质有36个基因表达值gi,其中i={1,2...36}。为了减少复杂性,通过下面等式(1)中给出的三个周期的平均值来计算跨越12个时间点的每个时刻的表达值。
[0067][0068]
公式(1)中,gi代表的是在i时刻的基因表达值,借助公式(1)我们可以计算出某种蛋白质在12个时间点的基因表达值。
[0069]
基因时序表达数据是在感兴趣的生物学过程中的一系列时间点收集的,从而反映了该过程中基因的动态活性。目前,现有的一些识别方法使用阈值来确定基因在某个时间点是否表达,因此我们使用3σ法则来计算活动阈值。设g i
表示基因g在时间点i的表达值,则通过以下公式计算可以得到活动阈值。
[0070][0071][0072][0073]
tg=ug+3ρg(1-fg)
ꢀꢀꢀ
(5)
[0074]
其中n代表基因表达谱中时间点的数目,公式(2)计算的是基因表达值的平均值,公式(3)计算的是基因表达值的标准差的值,公式(4)计算的是基因表达值的波动性值,公式(5)就是我们用来计算蛋白质在时间点i是否活跃的阈值。如果某个蛋白质在某个时间点的基因表达值gi大于等于这个阈值tg,我们就认为在该时刻这个蛋白质是处于活跃的状态,如果两个蛋白质在某个时间点都处于活跃状态,并且它们在静态ppi网络中有相互作用,那么在这个时间点的动态子网络中,这两个相互活跃的蛋白质之间就一定存在着一条边,一
般来说,这种相互作用被认为是一种动态的相互作用。至此我们已经把静态ppi网络转化为12个动态ppi子网络。
[0075]
步骤3:对于任一子网中的任一节点u来说,计算出该节点的点边缘聚类系数值decc(u,v),最后求该节点与其所有邻居之间的点聚类系数值之和sum_decc(u);其中decc(u,v)、sum_decc(u)的计算公如下(6)、(7)表示:
[0076][0077][0078][0079]
其中在公式(6)中,cn
u,v
表示结点u,v共同邻居的个数,ku,kv分别表示结点u,v的度,dccu,dccv表示结点u,v的点聚集系数,其计算如上述公式(8)所示,在公式(8)中kv表示结点v的度,nv表示由结点v的邻居结点之间组成的边数目。最后借助公式(7)对节点u和所有邻居节点v之间decc(u,v)值求和,其中v∈nu表示与u相连的所有邻居节点的集合。
[0080]
步骤4:对于任一子网中的任一节点u来说,计算出该节点与邻居节点之间的基因共表达值pcc(u,v),最后求该节点与其所有邻居之间的共表达系数之和sum_pcc(u);其中两个蛋白质节点之间的共表达系数pcc(u,v)值计算如公式(9)所示,sum_pcc(u)计算如公式(10)所示:
[0081][0082][0083]
在公式(9)中,u={u1,u2...un},v={v1,v2...vn}分别表示的是蛋白质u和蛋白质v的n个基因表达值。u’和v’分别表示其基因表达值的平均值。如果两个相互作用的蛋白质u,v基因的共表达程度越高,计算出来的pcc(u,v)值也就越大。在公式(10)中,v∈nu表示与u相连的所有邻居节点的集合。
[0084]
步骤5:对于任一子网中的任一节点u来说,计算出该节点和邻居之间的亚细胞定位得分值sls(u,v),最后求该节点与其邻居之间的亚细胞定位得分值之和sum_sls(u);其中蛋白质节点的亚细胞定位得分sls(u,v)值计算如公式(11)所示,sum_sls(u)计算如公式(12)所示:
[0085][0086]
[0087]
其中在公式(10)中|n|、|m|表示节点u、v在细胞核中出现的次数,在公式(11)中v∈nu表示与u相连的所有邻居节点的集合。
[0088]
步骤6:对于任一子网中的任一节点u来说,计算出该节点与邻居节点之间的go注释值go_sim(u,v),最后求该节点与其所有邻居之间的共表达系数之和sum_go(u);其中蛋白质节点之间的go注释相似度值go_sim(u,v)计算如公式(13)所示,sum_go(u)计算如公式(14)所示:
[0089][0090][0091]
其中在公式(13)中,分子中的绝对值表示蛋白质u和v具有相同的go注释的数量。分母|gou|和|gov|表示蛋白质u和v拥有go注释的数量。在公式(14)中,v∈nu表示与u相连的所有邻居节点的集合。
[0092]
步骤7:对于每一个节点u来说,将上述的属性值进行相加,得到蛋白质u的最终关键性得分值ess_pro(u);最后将ess_pro(u)的值从大到小排序。其中蛋白质节点的关键性值如下公式(15)所示:
[0093]
ess_pro(u)=sum_pcc(u)+sum_go(u)+sum_sls(u)+sum_decc(u)
ꢀꢀꢀ
(15)
[0094]
其中在公式(15)中的sum_pcc(u)、sum_go(u)、sum_sls(u)、sum_decc(u)分别是上述步骤(3)-(6)中所求的值。
[0095]
步骤8:最后将蛋白质节点按ess_pro(u)的值从大到小排序输出。就是将步骤7中所求的蛋白质关键性值进行降序排序,然后从大到小将蛋白质节点输出,这就是最终的实验结果。
[0096]
实施例:
[0097]
我们分别在mips、krogan、dip三个数据集上对我们提出的算法fmsbi进行了试验。表1给出了gavin、krogan、dip三个数据集的详细信息,包括了每个网络包含的蛋白质数量及蛋白质之间相互作用的数量。表2给出的是蛋白质生物属性数据集的信息。
[0098]
表1蛋白质相互作用网络数据集
[0099]
ppi数据集蛋白质数量相互作用数量mips454612319krogan26747075dip509324743
[0100]
表2蛋白质生物属性数据集
[0101]
生物数据集备注基因表达谱集版本:gse3431,每个基因包括36个时间点的表达值亚细胞定位集亚细胞定位集包含2332个位置得分go注释集go注释包括了7014个蛋白质的go注释信息
[0102]
为了评价fmsbi方法在关键蛋白质预测方面的性能,我们将其与其他关键蛋白质
识别方法分别进行比较,我们引入统计学性能评估方法,包含六个评价指标:敏感性(sn)、特异性(sp)、阳性预测值(ppv)、阴性预测值(npv)、f值和准确率(acc)。这些统计指标的定义分别如下:
[0103][0104][0105][0106][0107][0108][0109]
其中tp表示为预测为关键蛋白质的关键蛋白质数量,fn被预测为非关键蛋白质的关键蛋白质数量;tn被预测为非关键蛋白质的非关键蛋白质数量,fp被预测为关键蛋白质的非关键蛋白质数量。
[0110]
fmsbi算法与其他算法在dip、mips、krogan三个数据集上关于六个评价指标(sn,sp,ppv,npv,f,acc)的对比实验结果如表3、表4、表5所示。从表3、表4、表5中可看出,在dip这一ppi数据集上,本发明提出的算法fmsbi在识别关键蛋白质的性能上优于dc、ec、bc、lac、pec、wdc、udonc、lbcc等对比算法,表明本明提出的方法具有一定的优越性。而在mips、krogan两个ppi数据集上,本发明提出的算法fmsbi在识别关键蛋白质的性能上也优于dc、ec、sc、ic、nc、lac、pec、wdc等对比算法,更加表明本明提出的方法具有一定的优越性。
[0111]
表3发明与其他算法在dip数据集上的六个指标的对比图
[0112][0113]
表4发明与其他算法在dip数据集上的六个指标的对比图
[0114][0115]
表5本发明与其他算法在krogan数据集上的六个指标的对比图
[0116][0117]
从表3、表4中可看出,在dip这一ppi数据集上,本发明提出的算法fmsbi在识别关键蛋白质的性能上优于dc、ec、bc、lac、pec、wdc、udonc、lbcc等对比算法,表明本明提出的方法具有一定的优越性。表5中,在mips、krogan两个ppi数据集上,本发明提出的算法fmsbi在识别关键蛋白质的性能上也优于dc、ec、sc、ic、nc、lac、pec、wdc等对比算法,更加表明本明提出的方法具有一定的优越性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1