用于检测胎儿亚染色体畸变的母体血浆的组合的基于尺寸和基于计数的分析
用于检测胎儿亚染色体畸变的母体血浆的组合的基于尺寸和基于计数的分析
1.相关申请的交叉参考
2.本技术是申请号为201680006999.8的中国专利申请的分案申请,并且要求2015年 1月23日提交的题为“用于检测胎儿亚染色体畸变的母体血浆的组合的基于尺寸和计数的分析(combined size-and count-based analysis of maternal plasma for detection offetal subchromosomal aberrations)”的美国临时申请号62/107,227的优先权,其全部内容出于所有目的以引用的方式并入文中。本技术也涉及2008年7月23日提交的lo等人的题为“使用大规模平行基因组测序诊断胎儿染色体非整倍性(diagnosing fetalchromosomal aneuploidy using massively parallel genomic sequencing)”的美国专利公布 2009/0029377;以及2010年11月5日提交的lo等人的题为“基于尺寸的基因组分析 (size-based genomic analysis)”的美国专利号8,620,593,其全部内容出于所有目的以引用的方式并入。
背景技术:
3.母体血浆中的无细胞dna包含胎儿dna和母体dna的混合物。母体血浆的非侵入式产前测量可用于通过从亚染色体区域计数dna片段来检测亚染色体拷贝数畸变 (cna)。但是,计数并不将胎儿dna与母体dna区分开。因此,通过计数dna片段检测的畸变可以从胎儿或母亲得到。因此,当一个母亲自身是cna的载体时,无法辨别她的胎儿是否遗传了cna。此外,当分析更多的亚染色体区域时,假阳性结果将变得更加普遍。
4.实施例可以解决这些和其它问题。
技术实现要素:
5.实施例使用结合基于计数和基于尺寸的母体样本分析(包括母体dna和胎儿dna) 以检测胎儿亚染色体拷贝数畸变(cna)的策略。可以使用基于计数的分析来检测区域中的cna。dna分子的基于尺寸的分析也可以用于分析确定为具有cna的区域,其中基于尺寸的分析可用于将源于胎儿或母体,或源于两者的畸变区分开。
6.其它实施例涉及与本文所述的方法相关的系统和计算机可读介质。
7.参考以下具体实施方式和附图,可以更好地理解本发明实施例的性质和优点。
附图说明
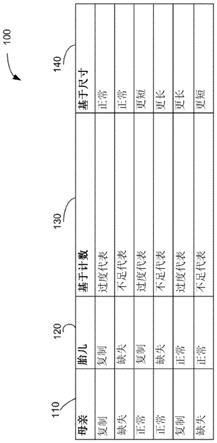
8.图1显示了根据本发明的实施例的用于基于计数和基于尺寸的结果的组合的六个情形的表100。
9.图2显示了根据本发明的实施例的在胎儿的染色体2上的区域中具有拷贝数增益的病例的组合的基于计数和基于尺寸的分析的示例性处理流程。
10.图3是根据本发明的实施例的通过分析来自怀有胎儿的雌性受试者的生物样本来识别胎儿的胎儿基因组中的亚染色体畸变的方法300的流程图。
11.图4显示了根据本发明的实施例的用于基于计数和基于尺寸的结果以及f
cna
值的组合的六个情形的表400。
12.图5是显示六个母体血浆dna样本的基于计数的分数和基于尺寸的分数以说明本发明的实施例的准确度的表500。
13.图6是显示关于来自胎儿或母亲或两者的cna的6个病例的信息的表600。
14.图7显示根据本发明的实施例的表600中六个病例的母体血浆dna的组合的基于计数和基于尺寸的分析。
15.图8是显示根据本发明的实施例的在每个病例中没有可检测的cna的测试区域的基于计数和基于尺寸的z分数的表800。
16.图9是显示根据本发明的实施例的通过分割方法确定的由100-kb单元组成的扩增区域的图900。
17.图10是显示根据本发明的实施例的通过分割方法确定的由100-kb单元组成的缺失区域的图1000。
18.图11显示了可用于根据本发明的实施例的系统和方法的示例性计算机系统10的框图。
具体实施方式
19.术语
20.本文所用的术语“生物样本”是指从受试者(例如人,例如孕妇)取得并含有一种或多种所感兴趣的核酸分子的任何样本。实例包括血浆、唾液、胸膜液、汗液、腹水、胆汁、尿液、血清、胰液、粪便和子宫颈涂片样本
21.术语“核酸”或“多核苷酸”是指单链或双链形式的脱氧核糖核酸(dna)及其聚合物。除非具体地限定,否则该术语包括含有天然核苷酸的已知类似物的核酸,其具有与参考核酸相似的结合特性并且以类似于天然存在的核苷酸的方式进行代谢。除非另有说明,特定的核酸序列也隐含地涵盖其保守修饰的变体(例如,简并密码子取代)、等位基因、直向同源物、单核苷酸多态性(snp)和互补序列以及明确指出的序列。具体地,简并密码子取代可以通过产生其中一个或多个所选(或全部)密码子的第三位置被混合碱基和/或脱氧肌苷残基取代的序列来实现(batzer ma等人,nucleic acid res 1991;19:5081; ohtsuka e等人,j biol chem 1985;260:2605-2608;和rossolini gm等人,mol cell probes1994;8:91-98)。
22.术语“序列读取”是指从全部或部分核酸分子获得的序列,例如dna片段。在一个实施例中,仅对片段的一端进行测序,例如约30个碱基。然后将测序的读数与参考基因组对比。或者,可以对片段的两端进行测序以产生两个测序读数,其可以提供更高的比对准确度并且还提供该片段的长度。在又另一个实施例中,线性dna片段可以例如通过连接进行环化,并且可以对跨越连接位点的部分进行测序。
23.术语分数胎儿dna浓度可与术语胎儿dna比例和胎儿dna部分互换使用,并且是指存在于从胎儿得到的母体血浆或血清样本中的dna分子的比例(lo ymd等人,amj hum genet 1998;62:768-775;lun fmf等人,clin chem 2008;54:1664-1672)。
24.术语“尺寸分布”通常涉及生物样本中dna片段的尺寸。尺寸分布可以是提供多种
尺寸的dna片段的分布的直方图。可使用各种统计参数(也称为尺寸参数或只是参数) 来将一个尺寸分布与另一个尺寸分布区分开。一个参数是特定尺寸或尺寸范围的dna 片段相对于所有dna片段或相对于另一个尺寸或范围的dna片段的百分比。
25.本文所用的术语“参数”意指表征定量数据集的数值和/或定量数据集之间的数值关系。例如,第一量的第一核酸序列与第二量的第二核酸序列之间的比例(或比例的函数) 是参数。
26.本文所用的术语“分类”是指与样本的特定性质相关的任何数字或其它字符(包括单词)。例如,“+”符号可以表示样本被分类为具有缺失或扩增(例如,重复)。术语“截止”和“阈值”是指操作中使用的预定数字。例如,截止尺寸可以指片段大于该尺寸被排除的尺寸。阈值可以是特定分类应用的高于或低于该值的值。这些术语中的任一个可以在这些上下文中使用。
[0027]“亚染色体区域”是小于染色体的区域。亚染色体区域的实例是尺寸为100kb、200 kb、500kb、1mb、2mb、5mb或10mb的那些。亚染色体区域的另一个实例是对应于染色体的一个或多个条带或子带或一个臂的区域。条带或子带是在细胞遗传学分析中观察到的特征。亚染色体区域可以通过其与参考人类基因组序列相关的基因组坐标来表示。
[0028]
基于母体血浆dna的非侵入性产前检测已经扩展到包括检测某些亚染色体拷贝数畸变(cna),也称为拷贝数畸变(cna)。但是,假阳性结果是普遍的,特别是在分析更多亚染色体区域时。虽然检测率高,假阳性率低,但是使用母体血浆中的无细胞dna 用于胎儿亚染色体非整倍体的非侵入性产前检测(nipt)由于阳性预测值不够高而目前尚未广泛用作筛选试验。
[0029]
以下描述表明,基于尺寸的分析可以用作独立的方法来验证由基于计数的分析检测的cna。另外,显示出,基于尺寸和基于计数的分析的组合可以确定胎儿是否已经从其母亲遗传了can,母亲本身是cna的载体。使用基于尺寸和基于计数的分析的组合的实施例可以区分通过分析母体生物样本(例如,使用测序和测序结果的分析)检测到的畸变的来源,即胎儿、母体或两者。该策略提高了当前测试的特异性。结果表明,实施例通过能够识别cna的来源提供了改进,而这是单独地仅使用基于计数的技术或基于尺寸的技术不可能达到的。
[0030]
i.介绍
[0031]
母体血浆中的无细胞dna包含胎儿dna和母体dna的混合物。在(nipt)中,母体血浆中无细胞dna的大规模平行测序(mps)用于胎儿染色体非整倍体的非侵入性产前检测(nipt)已被产前保健广泛采用(1,2)。这些方法是基于对映射到基因组的不同区域的母体血浆中dna片段的计数并因此被称为“计数方法”(3)。最近的研究已表明,这种方法可以通过使用更高的测序深度和适当的生物信息学分析来检测胎儿亚染色体异常(4-8)。事实上,一些公司开始为nipt提供许多临床上重要且相对普遍的亚染色体异常,例如digeorge综合症、cri-du-chat综合症、prader-willi/angelman综合症和1p36缺失综合症(9)。
[0032]
计数方法列举了母体样本中的胎儿和母体dna分子。其比较孕妇血浆中特定基因组区域与怀有正常胎儿的一组健康孕妇中相应值的相对表示。因此,基于计数的方法的异常结果可能由多于一种临床情况导致,即(i)胎儿、(ii)母亲或(iii)两者中存在拷贝数畸变(cna)(8,10)。如本文所使用的,母亲可以指生物母亲或替代物。术语怀孕的雌性受试者
也指两者。
[0033]
因此,如果母亲携带cna,则无法辨别胎儿是否遗传了畸变。实际上,母体拷贝数变异的存在是所报道的混淆nipt结果的原因之一(11)。snyder等人在两个病例中证明不一致的nipt结果可能归因于母体拷贝数变异的存在(11)。在一项最近的研究中,yin 等人报道了,55例样本中有35例存在母体拷贝数变异(63.7%),其中假阳性nipt结果为1,456例样本(12)。基于这一发现,yin等人建议对母体dna进行随访测试以排除病例中胎儿亚染色体畸变具有阳性nipt结果的母体拷贝数变异。因此,母体拷贝数变异的存在导致使用胎儿和母体dna的混合物检测胎儿亚染色体cna的不准确。
[0034]
最近,包括本公开的发明人的组开发了利用母体血浆中胎儿和母体dna分子之间的尺寸差异来检测胎儿非整倍体的方法(13)。来自胎儿的dna分子(也称为片段)具有与来自母亲的那些dna分子相比更短的尺寸分布(14,15)。因此,额外的胎儿染色体在胎儿三体性中的存在将缩短来自该染色体的母体血浆中dna的尺寸分布。这种基于尺寸的方法检测到来自血浆中非整倍体染色体的短片段的比例增加。这种方法允许以高准确度检测多种类型的胎儿全染色体非整倍体,包括三体性21、18、13和单体性x (13)。当存在母体拷贝数变异时,这种基于尺寸的方法的独立使用也苦于使用胎儿和母体dna的混合物检测胎儿亚染色体cna的不准确性。
[0035]
本公开显示,基于尺寸和基于计数的分析的组合被证实能够区分由母体血浆dna 测序检测到的畸变的来源,即胎儿、母体或两者。如果胎儿和母亲在特定亚染色体区域都有cna,当与没有任何cna的另一个亚染色体区域相比时,该区域的尺寸分布将不会有净差。另一方面,当与特定亚染色体区域中的母体dna相比时,例如当(i)胎儿在母体正常时具有微重复;或(ii)母亲在胎儿正常时具有微缺失时,如果胎儿dna存在相对过度表达,那么整体尺寸分布会缩短。相反,当与特定亚染色体区域中的母体 dna相比时,例如当(i)胎儿在母体正常时具有微缺失;或(ii)母亲在胎儿正常时具有微重复时,如果胎儿dna的代表性不足,则整体尺寸分布会拉长。以这种方式,基于尺寸的方法当与基于计数的方法组合时可以用于确定亚染色体cna的来源。
[0036]
ii.独立分析
[0037]
为了识别亚染色体cna,可以将整个基因组分为亚染色体区域(也称为单元)。在一些实施例中,单元可以小于其中具有cna的亚染色体区域可以包括多个单元的区域。具有畸变的连续单元可以限定具有cna的区域。在其它实施例中,区域可以对应于一个单元。如稍后更详细地解释,可以将单元合并以识别具有cna的区域(段)。
[0038]
这些单元可以具有例如约100kb、200kb、500kb、1mb、2mb、5mb或10mb 的尺寸。亚染色体区域也可以包括条带、子带和臂。在一个实施中,可以使用2,687个 1-mb单元。可以排除基因组的某些部分,例如重复区域。可以使用基于计数的方法和基于尺寸的方法来分析这些亚染色体区域,以确定该区域是否包括扩增或缺失。畸变可能不对应于整个区域,但是可以测试区域以识别是否在该区域的某处发生畸变。
[0039]
作为分析的一部分,分析母体样本中的无细胞dna片段以确定dna片段在基因组中的位置,例如相对于参考基因组。例如,可以对无细胞dna片段进行测序以获得序列读数,并且可以将序列读数映射(对齐)到参考基因组。如果生物是人类,则参考基因组将是潜在地来自特定亚群体的参考人类基因组。作为另一个实例,可以使用不同的探针(例如,在pcr或
其它扩增后)分析无细胞dna片段,其中每个探针对应于不同的基因组位置。在一些实施例中,无细胞dna片段的分析可以通过接收对应于无细胞 dna片段的序列读数或其它实验数据,然后使用计算机系统分析实验数据来进行。
[0040]
a.基于计数的分析
[0041]
为了进行基于计数的分析,实施例可以计数来自位于每个区域中的母体样本的dna 片段的数量。可将一个区域中的dna片段的数目与一个或多个计数阈值进行比较,以确定该区域是否呈现cna。计数阈值可以基于健康对照区域中的相应计数来确定以区分 cna和没有cna的区域。如果数字高于高阈值,则识别扩增。如果数字低于低阈值,则可以识别缺失。本领域技术人员将知道如何确定这样的阈值。
[0042]
将dna片段的数目标准化,使得可以在不同受试者之间进行比较,其中可以分析不同量的dna片段。标准化可以用许多方式进行,例如通过将一个区域的相应数目除以一个或多个其它区域(潜在地整个基因组)的相应数目的总和。与一个或多个其它区域的相应数目的这种比较也可以总是通过分析样本间相同数目的dna片段来完成,这使得总和总是相同的。因此,可以直接使用一个区域的计数数目,例如,因为总和可以有效地包括在阈值中。因此,在这样的一个实施例中,仍然实施数目的比例。因此,实施例可以从第一区域的第一个数目和第二区域的第二个数目计算计数参数。
[0043]
区域中的标准化数目的dna片段也可以称为基因组代表(gr)。标准化通过与不同区域的第二数目的dna片段的比率进行。例如,一个区域的gr可以是位于该区域中的dna片段的数目除以分析中使用的所有dna片段。一个区域的gr可以对应任何量,例如dna片段的数目、dna片段重叠的碱基数、或区域中dna片段的其它量度。
[0044]
在一些实施例中,计数参数可以对应于分数,例如以下列方式。可以确定对照的测试区域的基因组代表(gr)的平均值和标准偏差(sd)。可以使用以下等式(7),计算每个样本的测试区域的基于计数的z分数:
[0045][0046]
在文中的实例中,将》3和《-3的基于计数的z分数分别用作指示拷贝数增益和拷贝数丢失的计数阈值。在这样的一个实例中,z分数可以对应于与3或-3进行比较的计数参数。可以使用其它计数阈值,例如除3外的值。在其它实例中,gr
sample
是计数参数并且其它项可以移到等式的左侧以用作计数阈值的一部分。基于计数的方法的更多细节可以在美国专利公布2009/0029377中找到。
[0047]
b.基于尺寸的分析
[0048]
如上所述,胎儿dna片段小于母体dna片段。这种尺寸上的差异可用于检测胎儿中的cna。如果胎儿在第一区域具有扩增,则该区域的dna片段的平均尺寸将低于不具有扩增的第二区域;第一区域中的额外的较小的胎儿dna将减少平均尺寸。类似地,对于缺失,一个区域的较少的胎儿片段将导致大于正常区域的平均尺寸。尺寸的实例包括长度或质量。
[0049]
可以使用其它统计值,例如给定尺寸的累积频率或不同尺寸的dna片段的量的各种比例。累积频率可以对应于给定尺寸或更小尺寸的dna片段的比例。统计值提供关于dna片段的尺寸分布的信息,以与健康对照受试者的一个或多个尺寸阈值进行比较。与计数阈值一样,本领域技术人员将知道如何确定该阈值。
[0050]
因此,为了进行基于尺寸的分析,实施例可以计算位于第一亚染色体区域中的核酸分子的尺寸的第一统计值,并计算位于参考区域中的核酸分子的尺寸的参考统计值。可以在第一统计值与参考统计值之间确定分离值(例如差值或比例)。分离值也可以由其它值确定。例如,可以由多个区域的统计值确定参考值。可以将分离值与尺寸阈值进行比较以获得尺寸分类(例如,dna片段是否更短、更长或与正常区域相同)。
[0051]
一些实施例可以计算每个测试区域的参数(分离值),其被定义为使用以下等式的测试区域与参考区域之间的短dna片段的比例差(13):
[0052]
δf=p(≤150bp)
test-p(≤150bp)
ref
[0053]
其中,p(≤150bp)
test
表示来源于尺寸≤150bp的测试区域的测序片段的比例, p(≤150bp)
ref
表示来源于尺寸≤150bp的参考区域的测序片段的比例。在其它实施例中,可以使用其它尺寸阈值,例如但不限于100bp、110bp、120bp、130bp、140bp、160bp 和166bp。在其它实施例中,尺寸阈值可以用碱基或核苷酸或其它单位表达。在一些实施中,参考区域可以被定义为除测试区域外的所有亚染色体区域。在其它实施中,参考区域可以仅仅是除测试区域外的亚染色体区域的一部分。
[0054]
在基于计数的分析中使用的相同的对照组可用于基于尺寸的分析。可以使用对照的δf的平均值和sd值计算测试区域的基于尺寸的z分数(13)。
[0055][0056]
在一些实施例中,》3的基于尺寸的z分数表示测试区域的短片段的比例增加,而《-3 的基于尺寸的z分数表示测试区域的短片段的比例减少。可以使用其它尺寸阈值。基于尺寸的方法的进一步细节可以在美国专利号8,620,593中找到。
[0057]
为了测定dna片段的尺寸,至少一些实施例可以与其中可以分析染色体来源和分子长度的任何单个分子分析平台一起使用,例如电泳、光学方法(例如光学映射及其变体, en.wikipedia.org/wiki/optical_mapping#cite_note-nanocoding-3和jo等人,proc natl acad sciusa 2007;104:2673-2678)、基于荧光的方法、基于探针的方法、数字pcr(基于微流体或基于乳液,例如beaming(dressman等人,proc natl acad sci usa 2003;100:8817-8822)、 raindance(www.raindancetech.com/technology/pcr-genomics-research.asp))、滚环扩增、质谱、熔解分析(或熔解曲线分析)、分子筛等。作为质谱的一个实例,较长的分子将具有更大的质量(尺寸值的一个实例)。
[0058]
在一个实例中,dna分子可以使用双端测序方案进行随机测序。两端的两个读数可以被映射(对齐)到参考基因组,其可能被重复屏蔽。dna分子的尺寸可以由两个读取所映射的基因组位置之间的距离来确定。
[0059]
iii.组合的基于计数和尺寸的方法
[0060]
基于计数的方法比较特定基因组区域与一组怀有正常胎儿的健康孕妇的相对表示。因此,基于计数的方法的异常结果将通知胎儿或母体或两者都具有拷贝数畸变。另一方面,基于尺寸的方法是基于母体样本中dna分子的尺寸分布的差异,这取决于dna分子的来源。因此,与来自母亲的dna分子相比,来自胎儿的dna分子具有更短的尺寸分布。因此,如果胎儿和母亲在特定的亚染色体区域中具有拷贝数畸变,则当与不具有拷贝数畸变的另一个亚染色体区域比较时,在该区域中的尺寸分布将不存在净差。
[0061]
另一方面,当与特定的亚染色体区域中的母体dna比较时,例如当(i)胎儿具有微重复(或更大程度的微扩增),而母体是正常时;或(ii)母亲具有微缺失,而胎儿是正常时,如果胎儿dna存在过表达,则尺寸分布会缩短。相反,当与特定亚染色体区域中的母体dna比较时,例如当(i)胎儿具有微缺失,而母亲是正常时;或(ii)母亲具有微重复,而胎儿是正常时,如果胎儿dna的代表性不足,则尺寸分布会拉长。
[0062]
图1显示了根据本发明的实施例的用于基于计数和基于尺寸的结果的组合的六个情形的表100。在一些实施例中,正常的基于尺寸的分类对应于当统计值近似等于期望值 (例如参考控制)时。列110显示了母亲作为重复(或更大程度的扩增)、缺失或正常的各种状态。列120显示了胎儿作为重复(或更大程度的扩增)、缺失或正常的各种状态。列130显示了过度代表(对应于以上实例中的正基于计数的z分数)和代表性不足(对应于以上实例中的负基于计数的z分数)的基于计数的分类。列140显示了正常(两个区域中的相同尺寸)、较短(对应于以上实例中的正基于尺寸的z分数)和较长(对应于以上实例中的负基于尺寸的z分数)的基于尺寸的分类。
[0063]
观察表100,仅来自胎儿的cna的病例将具有在与基于计数的z分数相同方向上的基于尺寸的z分数。例如,基于计数的z分数的正值表示该区域的dna片段的过度代表,并且基于尺寸的z分数的正值表明该区域的dna片段较短,从而表明仅针对胎儿的扩增(例如,复制)。相反,基于计数的z分数和基于尺寸的z分数的负值分别表示该区域的代表性不足和更长的dna片段,从而表明仅针对胎儿的缺失。
[0064]
对于母亲携带cna的病例,基于尺寸的分析将用于确定胎儿是否遗传了母亲的畸变。胎儿遗传了来自母体的畸变的病例将具有在正常范围内的基于尺寸的z分数,因为受影响区域的胎儿和母亲dna的相对比例与其它基因组区域相比没有变化。例如,基于计数的分析的过度代表分类和基于尺寸的分析的正常分类显示胎儿遗传了扩增。而,基于计数的分析的代表性不足分类和基于尺寸的分析的正常分类表明胎儿遗传了缺失。
[0065]
另一方面,仅存在于母亲中的cna的病例将具有在与基于计数的z分数相反方向上的基于尺寸的z分数。因此,正基于计数的z分数(过度代表分类)和负基于尺寸的 z分数(较长分类)表明母体重复和胎儿的正常状态。相反,负基于计数的z分数(代表性不足分类)和正基于尺寸的z分数(较短分类)表明母体缺失和胎儿的正常状态。
[0066]
a.进行组合分析
[0067]
图2显示了根据本发明的实施例的在胎儿的染色体2上的区域中具有拷贝数增益的病例的组合的基于计数和基于尺寸的分析的示例性处理流程。对于染色体2,母体细胞显示为正常,并且对于区域202,胎儿细胞显示为具有重复。
[0068]
无细胞dna片段被显示存在于母体血浆205中。来自胎儿的dna分子(深红色片段220)具有比来自母亲的那些dna分子(黑色片段)更短的尺寸分布。从母体血浆中取出样本210。如图所示,进行双端测序以获得序列读数。可以使用双端测序(其包括测序整个dna片段)来确定dna片段的尺寸以及其位置,例如当序列读取被映射到参考基因组时。
[0069]
在基于计数的分析中,在框230处,计数与染色体2的相应单元232对齐的dna 片段。区域202的单元被识别为具有比正常区域204的单元更大的量。在该实例中,用于计数的单元小于用于分析z分数的区域。在其它实例中,可以对每个单元进行单独测定(因此,单元将与区域的尺寸相同)。在一些实施例中,可能需要多个连续单元以显示相同的畸变,例如
美国专利公布2014/0195164,其全文通过引用的方式并入。因此,即使正常区域204中的一个单元具有与区域202中两个单元相同的计数高度,该仓仍然包括在正常区域204中。
[0070]
在框240处,区域202的基因组代表(gr)被确定为区域202中的序列读取的计数数目除以序列读取的总计数。在其它实施例中,分母可以是仅一些单元的序列读数的计数。
[0071]
在框245处,确定基于计数的z分数。可以使用来自对照250的z分数值,例如以确定对照组的平均值和标准偏差(sd)。对照值可以用于对照受试者的相同测试区域或用于相似尺寸的其它区域。基于计数的z分数用向上箭头显示,表示大于计数阈值的正分数。
[0072]
在框260处,基于尺寸的分析可以接收表示过度代表的区域202的识别。基于尺寸的分析显示了区域202和参考区域的尺寸分布。如图所示,区域202的尺寸分布小于参考区域的尺寸分布。使用尺寸分布的统计值,可以在以下框中确定尺寸分布之间的这种关系的确定。
[0073]
在框270处,由p(≤150bp)
test
和p(≤150bp)
ref
确定分离值δf。其它统计值可用于其它实例。分离值显示为正值,因为区域202具有较高比例的150个碱基或更少的dna 片段。
[0074]
在框280处,确定基于尺寸的z分数。可以使用来自对照250的z分数值,例如以确定对照组的平均值和标准偏差(sd)。对照值可以用于对照受试者的相同测试区域或用于相似尺寸的其它区域。基于尺寸的z分数用向上箭头显示,表示大于尺寸阈值的正分数,其对应于小于参考区域的区域202的dna片段。
[0075]
根据图1的表100,过度代表的基于计数的分类和较短的基于尺寸的分类表示只有胎儿在区域202具有扩增。以这种方式,实施例可用以确定胎儿、母亲或两者是否具有识别的畸变。
[0076]
b.方法
[0077]
图3是根据本发明的实施例的通过分析来自怀有胎儿的雌性受试者的生物样本来识别胎儿的胎儿基因组中的亚染色体畸变的方法300的流程图。生物样本包括来自雌性受试者和胎儿的无细胞dna分子。方法300可以完全地或部分地用计算机系统进行。
[0078]
在框310处,测量生物样本中至少一些dna分子的尺寸。dna分子也称为片段,因为它们是整个基因组的片段,以及染色体的片段。尺寸可以通过任何合适的方法测量,例如上述方法。
[0079]
在框320处,识别在来自每个核酸分子的参考基因组中的位置。位置可以是基因组的任何部分,对于所提供的实施例,其是人类的,但可以是其它基因组。例如,位置可以是可以由基因组坐标(例如,特定坐标或坐标范围)定义的染色体的一部分。
[0080]
在一个实施例中,可以通过测序和比较序列信息与参考人类基因组序列来进行识别。在另一个实施例中,可以通过与已知染色体来源的一组探针杂交来进行该识别。探针可用一个或多个荧光标记,以微阵列形式或溶液标记。在又另一个实施例中,核酸分子可以由一组探针在溶液中或在固体表面上捕获,然后将捕获的(或剩余的未捕获的) 核酸分子进行测序。
[0081]
在框330处,使用基于计数的分析,例如,如第ii.a节所述,检测生物样本中第一亚染色体区域的畸变。例如,参考基因组可以分为两个单元,可以对映射到每个单元的 dna片段进行计数。基于计数,可以确定区域是否过度代表或代表性不足,作为检测畸变的一部分。如果确定既没有过度代表或代表性不足,则该区域可以被识别为正常。
[0082]
在一些实施例中,可以使用框320中确定的位置确定位于第一亚染色体区域中的第一量的dna分子。作为实例,第一量可以对应于完全位于第一亚染色体区域内、部分地位于第一亚染色体区域内的dna分子的数目以及dna分子与第一亚染色体区域重叠的基因组位置的数目。
[0083]
可以确定位于第二区域中的第二量的dna分子。在各种实施例中,第二区域可以是整个基因组、只有一个亚染色体区域、染色体(其可以包括第一亚染色体区域)和不相交的亚染色体区域,例如未测试的所有其它区域。可以由第一量和第二量计算计数参数。计数参数可以与一个或多个计数阈值进行比较,以确定存在于第一亚染色体区域的生物样本中的畸变类型的计数分类。
[0084]
畸变类型的实例是缺失、复制和更高阶扩增。每个畸变可以对应于不同的计数阈值。例如,当计数参数小于低阈值时,可以确定缺失,该低阈值将低于不存在畸变的区域的低阈值。当计数参数大于高阈值时,可以确定扩增,该高阈值将高于不存在畸变的区域的高阈值。如上所述,第二量可以包括在计数阈值中,其刚刚改变阈值,因此与由第一量和第二量确定计数参数相同。
[0085]
在框340处,使用基于尺寸的分析确定第一亚染色体区域的尺寸分类。尺寸分类可以表明位于第一亚染色体区域中的dna分子的尺寸分布是否比参考区域的尺寸分布更短、更长或相同。因为胎儿dna分子较小,所以尺寸分布的分析可以表明胎儿dna比例是否比参考区域更多(比参考更短的尺寸)或更少(比参考更大的尺寸),从而表明胎儿dna相对于参考区域是否存在过量、相同或不足。在各种实施例中,参考区域可以是整个基因组、只有一个亚染色体区域、染色体(其可以包括第一亚染色体区域)和不相交的亚染色体区域,例如未测试的所有其它区域。如以上针对图1所述,尺寸分类可用以区分胎儿和母亲之间畸变的不同可能性。
[0086]
在一些实施例中,可以计算位于第一亚染色体区域中的dna分子的尺寸的第一统计值。统计尺寸的实例是平均尺寸、尺寸分布的峰值尺寸、尺寸分布的模式、给定尺寸的累积频率等。可以确定位于参考区域中的dna分子的尺寸的参考统计值,以与第一统计值进行比较。可以确定第一统计值与参考统计值之间的分离值。分离值可以提供第一亚染色体区域中胎儿dna分子相对于参考区域的相对比例的量度。可以将分离值与一个或多个尺寸阈值进行比较以获得尺寸分类。
[0087]
在框350处,基于尺寸分类和计数分类,确定胎儿是否在第一亚染色体区域中具有畸变。可以使用图1进行确定,其中可以使用尺寸分类来区分每个用于过度代表和不足代表的计数分类的三种可能性。例如,当胎儿、母亲或两者都具有扩增时,可能发生过度代表的分类。
[0088]
如果只有胎儿具有扩增,则在第一亚染色体区域中比在参考区域中存在更大比例的胎儿dna分子,并且第一亚染色体区域的尺寸分布将更短。如果只有母亲具有扩增,则胎儿dna分子在第一亚染色体区域中比在参考区域中的比例更小,并且第一亚染色体区域的尺寸将更长。如果母亲和胎儿都具有扩增,则胎儿dna与母体dna的比例将与两者的高量相同,因此尺寸分布相同,导致正常分类。
[0089]
样本中存在的许多表观畸变将是非致病性拷贝数变异(cnv),其通常存在于人群中。因此,可以通过与各种数据库进行比较来进一步评分或排序样本中检测到的畸变。该数
据库具有关于cnv是否存在于特定人群中的感兴趣的区域中、cnv的类型(缺失或复制;增益或损失)、cnv的频率以及感兴趣区域中的致病性畸变是否报告的信息。例如,可以将从血浆dna识别的畸变的短列表与在1000个基因组 (http://www.1000genomes.org/)中识别的cnv、在变体数据库(dgv, http://dgv.tcag.ca/dgv/app/home)中策划的cnv和/或在decipher数据库 (decipher.sanger.ac.uk/)中记录的发育障碍所涉及的专家策划的微缺失和微复制综合症列表进行比较。
[0090]
在一个实施例中,与已知致病性畸变重叠的样本中识别的畸变将被分配较高的分数,而与已知非致病性cnv重叠的样本中识别的畸变将被分配较低的分数。每个畸变区域的分数可以组合以提供总致病分数。
[0091]
iv.含畸变分数
[0092]
将异常区域的基于计数的z分数的大小与携带畸变的血浆dna的比例相关联(17)。例如,如果只有胎儿具有畸变,则携带畸变的血浆dna的比例将与血浆样本中胎儿dna 的比例相关联。携带畸变的血浆dna的比例可用作额外的筛选来识别母亲具有畸变的病例。
[0093]
血浆中的含畸变分数(f
cna
,也称为acf)是指来自具有cna的细胞的血浆dna 的比例。理论上,如果只有胎儿携带畸变,只有那些胎儿得到的血浆dna分子会含有畸变;并且f
cna
将等于血浆中的胎儿dna分数。类似地,如果只有母亲携带畸变,只有那些母体得到的血浆dna分子才会含有畸变;f
cna
等于血浆中的母体dna分数。另一方面,如果母亲和胎儿都携带自然界中非马赛克的畸变(即,只有一些细胞携带畸变),则所有血浆dna分子将来自包含畸变的细胞;f
cna
将为100%。
[0094]
在一些实施例中,为了计算
fcna
,整个基因组可以分为2,687个1-mb单元,也称为单元。如上所述,可以计算每个单元的基于计数的z分数。在显示cna的区域中具有最高z分数的1-mb单元可用于计算f
cna
。如果该区域只有一个单元,那么将使用该单元。f
cna
可以如下计算(7):
[0095][0096]
gr
sample
是测试病例在受影响区域中具有最高z分数的1-mb单元的基因组代表,并且mean gr
control
是对照组中该单元的基因组代表的平均值。f
cna
是计数参数的另一个实例,其可以与计数阈值进行比较以确定是否存在畸变。但是,f
cna
也可以以其它方式使用。
[0097]
可以计算显示cna的每个区域的f
cna
。f
cna
可用于确定母亲中是否存在畸变。假设99%以上的母体血浆样本具有小于50%的胎儿dna分数(17,18),f
cna
》50%的病例表明母亲携带拷贝数畸变,因此不可能只是胎儿畸变。对于f
cna
《50%的病例,cna 潜在地存在于胎儿中。在一个实施例中,如果f
cna
小于50%(或其它截止值),则畸变被确定为胎儿或母亲中的马赛克。
[0098]
图4显示了根据本发明的实施例的用于基于计数和基于尺寸的结果以及f
cna
值的组合的六个情形的表400。表400与图1的表100类似。列410显示母亲的各种状态为正常、拷贝数增益(扩增)和拷贝数损失(缺失)。列420显示胎儿的各种状态。
[0099]
列430显示基于计数的分类。绿色向上指向的箭头表示大于高计数阈值(例如,》3) 的正基于计数的z分数。红色向下指向的箭头表示小于低计数阈值(例如,《-3)的负基于计
数的z分数。双箭头表示大量值的基于计数的z分数。大量值的基于计数的z分数可以对应于特定阈值的f
cna
,例如但不限于》40%、》45%、》50%、》55%和》60%。不同的截止值可用以定义“大量值的z分数”,例如但不限于对于正z分数而言》10、》15、》20、》25、》30、》35、》40、》45、》50、》55、》60以及对于负z分数而言 《-10、《-15、《-20、《-25、《-30、《-35、《-40、《-45、《-50、《-55。
[0100]
列440显示基于尺寸的分类。绿色向上指向的箭头表示大于高尺寸阈值(例如,》3) 的正基于尺寸的z分数。红色向下指向的箭头表示小于低计数阈值(例如,《-3)的负基于计数的z分数。
[0101]
列435显示了对于每个组合,f
cna
是否大于或小于50%。如所示,当f
cna
大于50%时,母亲会具有畸变。因此,可以计算f
cna
以确定母亲中是否存在畸变。f
cna
》50%表示母亲携带拷贝数畸变。在其它实施例中,可以使用f
cna
的其它阈值,例如但不限于》40%、》45%、》55%和》60%。
[0102]
因此,可以确定第一亚染色体区域的第一单元的第一单元计数参数(例如,gr
sample
)。第一亚染色体区域可以包括一个或多个单元。第一单元计数参数可以由位于第一单元中的dna分子的量确定,该量通过位于另一个区域中的dna分子的量进行标准化,该另一个区域可以是与用于第一亚染色体标准化的区域相同的区域或不同的区域。可以使用对照样本计算第一单元的对照单元计数参数的平均值(例如,mean gr
control
)。可以通过减去对照单元计数参数的平均值来获得结果并将结果除以对照单元计数参数的平均值 (例如,如上所示)来计算第一单元的第一分数(例如,f
cna
)。可以使用第一分数(例如,绝对值)是否大于截止值(对于上述实例为50%)来识别雌性受试者是否在第一亚染色体区域具有畸变。取决于第一分数的定义,例如是否使用2和100%的因子,可以使用其它截止值。
[0103]
v.结果
[0104]
下面的结果证实了实施例能够正确地识别检测到的亚染色体畸变是来自胎儿、母亲还是两者的能力。这样的结果表明较现有技术的改进,现有技术将所有亚染色体畸变错误分类为来自胎儿,导致假阳性。
[0105]
a.第一组结果
[0106]
使用尺寸剖析方法分析来自先前研究的具有已知胎儿微缺失和微重复的六个母体血浆dna样本的双端测序数据。在六例测试病例中,有5例涉及染色体3q、4q和22q 的胎儿得到的亚染色体缺失或重复,1例在22q上的母体遗传的微重复。每个测序的dna 片段的尺寸由双端读数的开始和结束坐标确定。
[0107]
对于每个测试案例,目标区域被定义为通过基于计数的分析识别的含有拷贝数畸变的区域。参考区域(方法300的第二区域)包含在不含畸变的常染色体上的所有未受影响的基因组区域。在先前研究中用于基于计数的分析的具有正常胎儿核型的8个单胎怀孕病例的相同组被应用于尺寸分析作为参考对照(7)。然后计算每个测试样本的目标区域的基于尺寸的z分数。
[0108]
图5是显示六个母体血浆dna样本的基于计数的分数和基于尺寸的分数以说明本发明的实施例的准确度的表500。基于计数的z分数显示畸变是存在的。基于计数的分数的范围对应于区域的1-mb单元。
[0109]
使用图4的表400,仅来自胎儿的拷贝数畸变的病例将具有在与基于计数的z分数
相同方向上的基于尺寸的z分数,即,正数将表明扩增,而负数将表明缺失。使用平均值的3个sd的z分数截止值,在病例01-04和06中,通过基于计数的方法检测到的所有拷贝数畸变被独立地确认为仅来自胎儿(如表500所示)。
[0110]
病例05是涉及胎儿的怀孕,该胎儿从其母亲遗传了在染色体22q上的2.4mb的微重复。因为母亲自身携带微复制,所以涉及到的三个1-mb单元的基于计数的z分数非常高(范围为39.7至71.7)。但是,这种分析本身并不表明胎儿是否从母亲遗传了微重复。
[0111]
使用基于尺寸的分析与基于计数的分析的组合,合并的3-mb单元显示基于尺寸的 z分数在正常范围内。当畸变来自于胎儿和母亲时,此观察结果与受影响区域中母体血浆dna的尺寸分布保持不变(因为来自胎儿和母亲的相对贡献没有改变)相一致。另一方面,如果胎儿没有从母亲遗传微重复,则受影响区域中的短片段的比例将会降低,导致与正基于计数的z分数相反的负基于尺寸的z分数。
[0112]
b.第二组结果
[0113]
图6是显示关于来自胎儿或母亲或两者的cna的6个病例的信息的表600。其中三个病例已包括在只评价基于计数的方法的先前研究中(7)。其余三例是以前未分析过的新病例。将具有正常胎儿和母体核型的单胎怀孕病例用作对照。因为三个新病例和已包括在先前研究中的三个病例均采用不同的文库制备试剂盒制备,所以这两组不同对照用于分析这两组测试病例。每组利用相同的文库制备试剂盒制备,并用与相应组的测试病例相同数量的泳道进行测序。
[0114]
列610显示母亲是否存在畸变的已知状态。列620显示了胎儿是否存在畸变的已知状态。为了测试实施例是否可以预测已知状态,对于每个病例,在四个目标区域上进行组合的基于计数的分析和基于尺寸的分析,四个目标区域包括在染色体4上的两个2-mb 区域、在染色体12上的一个4-mb区域、以及在染色体22上的一个3-mb区域。
[0115]
图7显示根据本发明的实施例的表600中六个病例的母体血浆dna的组合的基于计数和基于尺寸的分析。图7中的分数可以使用图4的表400来分析以预测畸变是否在胎儿、母亲或两者上。列710显示了母亲是否存在畸变的预测状态。列720显示了胎儿是否存在畸变的预测状态。列710和720中的预测对应于列610和620中的已知状态。
[0116]
对于cna仅存在于胎儿中的两个病例(m10219和hk310),基于尺寸的方法证实了由基于计数的方法检测到的畸变。对于母亲自身携带畸变的四种病例,实施例成功地推断出两个胎儿遗传了畸变,另外两个胎儿没有遗传畸变。在这个队列中没有观察到假阳性。
[0117]
对于m10219,以13.4的基于计数的z分数检测到在染色体22上的3mb微复制。对于hk310,以-8.2的基于计数的z分数在同一区域中检测到3mb微缺失。这两个病例的f
cna
分别为21.3%和15.1%。m10219和hk310的此区域的基于尺寸的z分数分别为6.9和-6.3,表明受影响区域在m10219中具有较短的尺寸分布,而在hk310中具有较长的尺寸分布。在这两个病例中,基于尺寸的z分数在与基于计数的z分数相同的方向上,表明胎儿是在母体血浆中检测到的畸变的唯一来源。这些结果与图6中两个病例的临床信息一致。
[0118]
对于m14-13489-f1,以93.8的基于计数的z分数在染色体4上检测到2-mb微复制。对于dna 11-04530,以-61.9的基于计数的z分数在染色体4上的另一个区域中检测到 2-mb微缺失。f
cna
分别为69.1和82.5。对于具有异常的基于计数的z分数的区域,对于m14-13489-f1而言,相应的基于尺寸的z分数为-3.6,以及对于dna 11-04530而言为5.1。因此,在这两
个病例中,基于尺寸的z分数在与基于计数的z分数相反的方向上,这表明畸变只存在于母亲中。这些结果与图6中的临床信息一致。这些结果显示了一个实例,其对将所有畸变分配给胎儿的当前技术做出改进。在这里,避免了这种假阳性。
[0119]
对于pw503和m11879,在染色体22上检测到3mb微复制(基于计数的z分数: 71.6),以及在染色体12上检测到4mb微复制(基于计数的z分数:154.5)。这两个病例的f
cna
分别为100%和99.6%。目标区域的基于尺寸的分析显示两个病例的基于尺寸的z分数在正常范围内(基于尺寸的z分数:pw503为0.9,m11879为0.0),表明在这两个病例中母亲和胎儿具有微复制。这些结果也与图6中的临床信息一致。
[0120]
pw503和m11879是涉及胎儿的妊娠,胎儿分别从其母亲遗传了在染色体22q上的2.4mb微重复和3.5mb微重复。因为母亲自身携带微复制,因此确定了非常高的基于计数的z分数。但是,基于计数的分析本身并没有揭示胎儿是否从母亲遗传了微重复。使用基于尺寸的方法,两个病例的3mb和4mb区域分别显示了基于尺寸的z分数在正常范围内。当畸变来自胎儿和母亲时,此观察结果与受影响区域中母体血浆dna的尺寸分布保持不变相一致,因为与其它未受影响的区域相比,来自胎儿和母亲的相对贡献在受影响的区域中没有改变。另一方面,如在m14-13489-f1中,当胎儿没有从母亲遗传微重复时,受影响区域中的短片段的比例将会降低,导致与正基于计数的z分数相反的负基于尺寸的z分数。
[0121]
图8是显示根据本发明的实施例的在每个病例中没有可检测的cna的测试区域的基于计数和基于尺寸的z分数的表800。对于每个病例,利用组合分析在其它测试区域中没有检测到畸变。仅使用基于计数的方法,以6.61的基于计数的z分数在 m14-13489-f1中的染色体22上的3mb区域中检测到过度代表。f
cna
为14.8%,基于尺寸的z分数为-0.82。因此,通过基于计数的分析检测到的畸变并没有通过基于尺寸的分析证实,并且该区域被分类为正常,这与阵列cgh分析一致。
[0122]
因此,f
cna
可以用于区分母亲和胎儿具有畸变的情况和基于计数的分析中的假阳性。在以上针对m14-13489-f1的实例中,f
cna
为14.8%,远低于50%。因此,母亲和胎儿都不可能表现畸变,这对应于基于计数的分析为正以及基于尺寸的分析显示正常。以这种方式,f
cna
可以用作进一步检查。因此,在一些实施例中,可以确定当第一分数小于截止值时,并且当:计数分类表示扩增,并且尺寸分类表示在第一亚染色体区域中不存在畸变,或者计数分类表示缺失,并且尺寸分类表示在第一亚染色体区域中不存在畸变时,在第一亚染色体区域中不存在畸变。
[0123]
以下列方式对第二组结果的上述数据进行采样和处理。经书面知情同意和机构伦理委员会的批准,从威尔斯亲王医院妇产科、香港广华医院和荷兰拉德堡德大学医学中心招募了单胎妊娠妇女。如前所述收集和处理母体外周血样本(16)。使用qiaamp dspdna血液微型试剂盒从血浆中提取dna(16)。
[0124]
以下列方式进行血浆dna测序。我们按照制造商的说明,使用kapa文库制备试剂盒(kapa biosystems)制备了新病例的dna文库。连接物连接的血浆dna通过12 周期pcr富集。每个文库在hiseq 1500或hiseq 2500测序仪(illumina)上利用流动池的两个泳道进行测序。我们进行了50个周期的双端测序。如前所述,对齐和过滤双端读数(13)。对齐后,每个测序的dna片段的尺寸由双端读取的开始和结束坐标确定。
[0125]
对于包括在先前研究中的三个病例,如下所述,对这些母体血浆dna样本的双端测
序数据进行重新分析。这些病例的血浆dna文库预先利用双端测序样本制备试剂盒 (illumina)制备,并在hiseq 2000测序仪(illumina)上利用流动池的一个泳道进行测序。
[0126]
vi.合并段
[0127]
如上所述,单元可以小于畸变区域,并且可以将显示畸变的连续单元组合以识别畸变区域。除了使用具有计数参数的连续单元,实施例可以使用例如二进制环形分割和隐马尔可夫模型的其它技术来识别对应于畸变区域的一组单元。可以将单元合并以形成对应于畸变区域的合并段。
[0128]
a.合并单元
[0129]
作为一个实例,使用特定尺寸的窗口将人类基因组分成非重叠的单元。窗口的尺寸的实例是10kb、50kb、100kb、500kb和1000kb等。在一些实施例中,滤出具有低映射性(例如小于10%)的单元。可以确定每个单元的dna分子的量,其中可以使用gc 校正来确定来自原始计数的量(chen ez等人,plos one.2011;6(7):e21791)。可映射性对应于通过对齐将源自区域的读取分配或识别回实际原始基因组位置的能力。一些区域具有低可映射性,例如,因为没有足够独特的核苷酸背景。这些区域在测序深度中的代表性不足。
[0130]
可以确定与单元i对齐的gc校正后的读取比例(称为基因组代表,gr),并称为 gri。gri可以进一步转化为测试样本zi的z分数统计量:
[0131][0132]
其中gri0和sdi0分别是携带整倍体胎儿的健康怀孕组(正常受试者)中对应于单元i的gr的平均值和标准偏差(sd)。
[0133]
然后可以将分割步骤沿着每个染色体应用于zi。该分割步骤可以将在相同方向上表现出基因组代表变化(例如,相对过度代表、相对不足代表、或没有变化)的连续单元合并成更大段,被称为合并段。分割可以以基因组坐标的升序或降序进行。可使用各种技术用于分割步骤。
[0134]
在一个实施例中,可以使用二进制环形分割和隐马尔可夫模型(https://www.bioconductor.org/packages/3.3/bioc/manuals/snapcgh/man/snapcgh.pdf)算法来实施该分割步骤。显示与由未受影响的对照或受试者建立的参考范围相比在统计学上显著升高的正z分数值的合并段可以被鉴定为候选微重复(或更一般地作为微扩增)。显示与由未受影响的对照或受试者建立的参考范围相比在统计学上显著降低的负z分数值的合并段可以被鉴定为候选微缺失。术语“候选”可以指该区域是胎儿畸变的候选者,其可以使用尺寸分析来证实。
[0135]
距正常范围的显著偏差可以通过不只是一个阈值来定义,例如,如方法300中所述。例如,可以分析合并段的尺寸以确定合并段是否大于长度阈值,例如至少3兆碱基(mb)。其它长度阈值的实例包括1mb、2mb、4mb、5mb、10mb等。
[0136]
并且,可以分析合并段的偏差的量值以确定量值是否超过偏差阈值。例如,合并段 (即包括合并段中的所有单元)的z分数绝对平均值可以被要求大于偏差阈值(例如, 1.5)。其它偏差阈值的实例包括1、2、3、5等。量值是计数参数的一个实例,或者可以被确定为计数参数与可以是参考范围的计数阈值的比较的一部分。z分数平均值可以是单独z分数的平均值或者使用整个合并段的dna分子的总量确定的z分数。
[0137]
在一些实施例中,可以进行单元的初步分析以识别可能形成满足长度阈值和/或偏差阈值的合并段的畸变单元。这样的初步分析也可以使用z分数分析。用于初步分析的阈值可以与用于整个合并段的偏差阈值不同(例如,较大)。一旦识别出彼此接近(例如,在指定长度内,例如不小于500kb的间隙)的足够数量的畸变单元,则可以在该区域中的单元上使用分割方法来识别合适的区域。然后,可以例如使用长度阈值和/或偏差阈值来分析该区域。可以单独地测试长度或偏差以识别候选者,或者可以需要两者以得到满足。
[0138]
b.含畸变分数(acf)
[0139]
如上所述,可以将含畸变分数用作计数参数,以确定存在于第一亚染色体区域的生物样本中的畸变类型的计数分类。因此,可以使用含畸变分数而不是区域的z分数或者例如通过合并段所定义的区域的单元的平均z分数。因此,含畸变分数可以用作距合并段的参考范围的偏差。含畸变分数可以对应于样本中含畸变的等效细胞的比例。
[0140]
可以以各种方式定义含畸变分数,例如使用第iv节中的定义,将其表示为f
cna
。但是,可以以其它方式定义含畸变分数,这里我们表示为acf。
[0141]
在一个实施例中,acf可以使用以下等式定义:
[0142][0143]
其中gr'是测试样本中显示微缺失或微重复的合并段(区域)的gr,gr0是对照(参考)样本中合并段的平均gr。
[0144]
在另一个实施例中,acf可以使用以下等式定义:
[0145]
or acf=|z
′×
cv0|
×2[0146]
其中z'是测试样本中显示微缺失或微重复的合并段(区域)的z分数。cv0是正常受试者(也称为对照或参考受试者或样本)中相应区域的变异系数(cv)。在一个实施例中,通过将测试样本中的区域的gr与正常受试者中对应区域的gr的平均值和标准偏差进行比较,例如根据第ii.a节中定义的z分数来重新计算合并段的z'。在另一个实施例中,也可以通过将100kb z分数的总和除以所涉及的单元数的平方根,由落入合并段中单元的一系列单独z分数来估计z'。
[0147]
acf可以反映出畸变的潜在来源组织。例如,如果畸变仅源于胎儿,则acf将等于胎儿dna分数。如果畸变仅源于母亲,则acf将远大于胎儿dna分数,因为一般而言,胎儿dna在血浆中占少部分比例。如果畸变源于两者,则分析的acf将接近100%。
[0148]
因此,可以使用acf与胎儿dna分数之间的分离值(例如,差值或比例)来分类在样本中看到的基因组畸变的来源组织。可以使用各种技术来计算样本的胎儿dna分数,例如基于snp、基于尺寸和基于chry的方法,例如,如美国专利公布2013/0237431 中所述。
[0149]
以下是如何使用acf与胎儿dna分数之间的差值来推断畸变是源自母亲还是胎儿的一些实例。例如,如果acf与胎儿dna分数之间的差值小于低阈值(例如2%,则畸变将被分类为“胎儿得到的畸变”。阈值的其它实例包括1%、3%、4%和5%。
[0150]
如果acf与胎儿dna分数之间的差值大于高阈值(例如,20%),则畸变将被分类为“涉及母亲的畸变”。高阈值的其它实例包括10%、30%、40%和50%。当超过高阈值时,畸变只会在母亲或母亲和胎儿两者中。
[0151]
因为来自背景母体细胞的畸变可以是马赛克(即,只有一部分(《100%)的贡献血
浆dna的母体细胞含有畸变),所以当畸变只源于母亲或母亲和胎儿两者时,acf与胎儿dna分数之间的差值可以具有近似值。在一个实施例中,只有显示acf超过一定阈值(例如,4%、5%、6%等)的区域可被认为是候选微缺失或微重复。
[0152]
第一亚染色体区域的第一单元的第一单元计数参数可以对应于整个区域的gr,例如当区域仅具有一个单元时。可以通过从第一单元计数参数中减去对照单元计数参数的平均值来确定该区域的第一分数(acf或f
cna
)。在各种实施例中,可以将减法的结果除以普通受试者的对照单元计数参数的平均值或标准偏差。
[0153]
如上所述,可以在生物样本中测量胎儿dna浓度。可以计算第一分数与胎儿dna 浓度之间的差值。确定第一分数是否大于截止值可以包括确定差值是否大于高阈值,例如,如上所述。此外,可以将差值与低阈值进行比较,以确定当差值低于低阈值时,只有胎儿在第一亚染色体区域具有畸变,例如如上所述。
[0154]
在第一亚染色体区域包括多个单元的实施例中,可以计算多个单元中每一个的相应分数。可以使用相应分数的总和来确定第一分数。例如,可以取平均值。作为另一个实例,第一分数是总和除以用于确定总和的单元数的平方根。
[0155]
c.结果
[0156]
图9是显示根据本发明的实施例的通过分割方法确定的由100-kb单元组成的扩增区域的图900。图900提供了微重复识别和综合解释的一个实例。将每个染色体分为100-kb 单元。在图900中,每个点表示100-kb单元中的z分数。
[0157]
进行二进制环形分割和基于隐马尔可夫模型(hmm)的分割。在染色体22中,两种分割算法都显示出一致的结果。使用》3mb的合并段尺寸的截止值以及》1.5的z分数平均值的量值的截止值,在阴影区域的面积中所示的合并段(chr22:17,000,000
–ꢀ
20,000,000)被识别为候选微复制。根据用于合并段的基于计数的合并z分数为72,含畸变分数(acf)被确定为100%。
[0158]
使用fetalquant算法,胎儿dna分数为22%(jiang p等人,bioinformatics.2012;28 (22):2883-90)。acf远大于胎儿dna分数,这表明母亲的畸变将存在于该区域中。对于合并区域,基于尺寸的z分数被确定为0.9,表明与3的截止值相比时正常的尺寸分布。因此,根据图1和4,尺寸分析表明,母亲和胎儿在该区域中将具有微重复。通过与数据库进行比较,发现该候选微复制与22q11复制综合症重叠。
[0159]
图10是显示根据本发明的实施例的通过分割方法确定的由100-kb单元组成的缺失区域的图1000。图1000提供了微缺失识别和综合解释的一个实例。将每个染色体分为 100-kb单元。在图1000中,每个点表示100-kb单元中的z分数。
[0160]
进行二进制环形分割和基于隐马尔可夫模型(hmm)的分割。在染色体4中,两种分割算法都显示出一致的结果。使用》3mb的合并段尺寸的截止值以及》1.5的z分数平均值的量值,在阴影区域的面积中所示的合并段(chr4:158,000,000-198,000,000)被识别为候选微缺失。根据用于合并段的基于计数的合并z分数为-74.5,含畸变分数(acf) 被确定为14.7%。
[0161]
使用fetalquant算法,胎儿dna分数为13%(jiang p等人,bioinformatics.2012;28 (22):2883-90)。acf非常接近于胎儿dna分数(例如,低于2%的低阈值),这表明只有胎儿在该区域中存在微缺失。对于合并段,基于尺寸的z分数被确定为-14,表明与-3 的截止
值相比时明显更长的尺寸分布。因此,根据图1和4,尺寸分析表明,胎儿在该区域中将具有微缺失。
[0162]
vii.概要
[0163]
虽然具有高检测率和低假阳性率,但是使用母体血浆中的无细胞dna的胎儿亚染色体非整倍体的nipt由于阳性预测值不足够高而目前不被广泛用作筛选试验。如果包括亚染色体cna,则该试验的阳性预测值被预期甚至更低,因为这些病症的个体成员甚至比全染色体非整倍体更罕见。此外,由于多个比较而导致假阳性的数量将随着更多测试目标而增加。据yin等人报道,其55例假阳性样本中有20例可能归因于测序和统计学错误(12)。如本发明的实施例所示,基于尺寸的分析可以作为确定通过基于计数的分析检测到的畸变的独立方法。结果表明,可以通过组合的基于计数和基于尺寸的方法,最大限度地减少由于统计误差导致的假阳性数量。
[0164]
在一些实施例中,为了实现在5%的胎儿dna分数下以95%灵敏度和99%特异性检测胎儿亚染色体cna的2-mb的分辨率,需要基于计数和基于尺寸的方法来分析约2 亿个分子(7)。另一方面,因为前三个月的胎儿dna分数中位数约为15%(17,19),所以可以使用约2000万个分子来实现相同的表现。该估计基于先前报道的数学关系,其中胎儿dna分数每增加两倍将导致相同测试性能所需分子的4倍降低(20)。因为相同的测序数据集可用于两种类型的分析,所以与需要用于单端测序的试剂的仅计数方案相比,实施例只需要用于双端测序的附加试剂成本。此外,两种方案所需的生物信息学处理的时间要求是可比的。
[0165]
总之,我们已经证明孕妇血浆dna的尺寸分析可以准确地检测胎儿亚染色体cna。基于尺寸和基于计数的方法的组合使用可以进一步确定胎儿、母亲或他们两者是否携带畸变。这种组合方法在帮助临床医生解释nipt的结果上非常有价值。
[0166]
viii.计算机系统
[0167]
本文提到的任何计算机系统可以利用任何合适数量的子系统。这种子系统的实例在图11中在计算机设备10中显示。在一些实施例中,计算机系统包括单个计算机设备,其中子系统可以是计算机设备的组件。在其它实施例中,计算机系统可以包括具有内部组件的多个计算机设备,每个是子系统。计算机系统可以包括台式和膝上型计算机、平板电脑、移动电话和其它移动装置。
[0168]
图11所示的子系统通过系统总线75相互连接。显示了耦合到显示适配器82的额外子系统,例如打印机74、键盘78、存储设备79、监视器76。耦合到i/o控制器71 的外围设备和输入/输出(i/o)装置可以通过本领域已知的任何数量的装置连接到计算机系统,例如输入/输出(i/o)端口77(例如,usb、)。例如,可以使用i/o 端口77或外部接口81(例如,以太网、wi-fi等)将计算机系统10连接到例如因特网、鼠标输入设备或扫描仪的广域网。通过系统总线75的互连允许中央处理器73以与每个子系统进行通信并控制来自系统存储器72或存储设备79(例如,固定盘,例如硬盘驱动器或光盘)的指令的执行,以及子系统之间的信息交换。系统存储器72和/或存储设备79可以包括计算机可读介质。另一个子系统是例如照相机、麦克风、加速度计等的数据采集装置85。本文提到的任何数据都可以从一个组件输出到另一个组件,并可以输出给用户。
[0169]
计算机系统可以包括多个相同的组件或子系统,例如通过外部接口81或内部接口连接在一起。在一些实施例中,计算机系统、子系统或设备可以通过网络进行通信。在这种
情况下,一台计算机可以被认为是客户机,另一台计算机可以被认为是服务器,其中每台计算机都可以是同一计算机系统的一部分。客户机和服务器每个可以包括多个系统、子系统或组件。
[0170]
应当理解,本发明的任何实施例可以以控制逻辑的形式使用硬件(例如,专用集成电路或现场可编程门阵列)和/或使用计算机软件经通常可编程处理器以模块化或集成的方式实施。如本文所用,处理器包括单核处理器、同一集成芯片上的多核处理器或单个电路板上或联网的多个处理单元。基于本文提供的公开内容和教示,本领域普通技术人员将知道并认识到使用硬件以及硬件和软件的组合来实现本发明的实施例的其它方式和/或方法。
[0171]
本技术中描述的任何软件组件或功能可以被实现为使用任何合适的计算机语言(比如,例如java、c、c++、c#、objective-c、swift或脚本语言,例如perl或python) 利用例如常规或面向对象的技术由处理器执行的软件代码。软件代码可以作为一系列指令或命令存储在用于存储和/或传送的计算机可读介质上,合适的介质包括随机存取存储器(ram)、只读存储器(rom)、例如硬驱动器或软盘的磁介质、或例如光盘(cd) 或dvd(数字通用盘)的光学介质、闪存等。计算机可读介质可以是这种存储或传送装置的任何组合。
[0172]
此类程序也可以使用适合于通过包括因特网在内的符合各种协议的有线、光学和/ 或无线网络进行传送的载波信号来编码和传送。因此,根据本发明的一个实施例的计算机可读介质可以使用利用程序编码的数据信号来创建。用程序代码编码的计算机可读介质可以与兼容装置一起打包或者与其它装置分开提供(例如,经由因特网下载)。任何这样的计算机可读介质可以驻留在单个计算机产品(例如硬盘驱动器、cd或整个计算机系统)上或内部,并且可以存在于系统或网络内的不同计算机产品上或内部。计算机系统可以包括监视器、打印机或其它合适的显示器以向用户提供本文提到的任何结果。
[0173]
本文所述的任何方法可以完全或部分地使用包括一个或多个处理器的计算机系统来执行,该计算机系统可被配置为执行这些步骤。因此,实施例可以针对被配置为执行本文描述的任何方法的步骤的计算机系统,潜在地具有执行相应步骤或相应的步骤组的不同组件。虽然以编号步骤表示,但是可以在同一时间或以不同的顺序执行本文的方法的步骤。此外,这些步骤的一部分可以与来自其它方法的其它步骤的一部分一起使用。并且,全部或部分步骤可以是任选的。另外,任何方法的任何步骤可以用模块、电路或用于执行这些步骤的其它器件来执行。
[0174]
在不脱离本发明的实施例的精神和范围的情况下,特定实施例的具体细节可以以任何合适的方式组合。但是,本发明的其它实施例可以针对涉及每个单独方面或这些单独方面的具体组合的具体实施例。
[0175]
为了说明和描述的目的,呈现了本发明的示例性实施例的上述描述。无意穷举或将本发明限制于所描述的精确形式,并且鉴于上述教示,许多修改和变化是可能的。
[0176]“一个”或“所述”的引述用以表示“一个或多个”,除非另有明确指出。除非另有明确指出,否则使用“或”用以意指“包容性或”,而不是“排他性或”。
[0177]
出于所有目的,本文提及的所有专利、专利申请、公布和说明书通过引用全文的方式并入。没有一个被认为是现有技术。
[0178]
本技术还涉及以下实施方案:
[0179]
实施方案1.一种通过分析来自怀有胎儿的雌性受试者的生物样本来识别胎儿的胎儿基因组中的亚染色体畸变的方法,所述生物样本包括来自所述雌性受试者和所述胎儿的无细胞dna分子,所述方法包括:
[0180]
对于所述生物样本中的多个dna分子中的每一个:
[0181]
测量所述dna分子的尺寸;
[0182]
识别所述dna分子在参考基因组中的位置;
[0183]
利用计算机系统通过以下检测所述生物样本中的第一亚染色体区域的畸变:
[0184]
确定位于所述第一亚染色体区域中的dna分子的第一量;
[0185]
确定位于第二区域中的dna分子的第二量;
[0186]
由所述第一量和所述第二量计算计数参数;以及
[0187]
将所述计数参数与一个或多个计数阈值进行比较以确定存在于所述生物样本中的所述第一亚染色体区域的畸变类型的计数分类;
[0188]
利用所述计算机系统通过以下确定所述第一亚染色体区域的尺寸分类:
[0189]
计算位于所述第一亚染色体区域中的dna分子的尺寸的第一统计值;
[0190]
计算位于参考区域中的dna分子的尺寸的参考统计值;
[0191]
确定所述第一统计值与所述参考统计值之间的分离值;以及
[0192]
将所述分离值与一个或多个尺寸阈值进行比较以获得所述尺寸分类;以及
[0193]
基于所述尺寸分类和所述计数分类来确定所述胎儿是否在所述第一亚染色体区域中具有畸变。
[0194]
实施方案2.根据实施方案1所述的方法,其进一步包括:
[0195]
通过以下识别所述第一染色体区域:
[0196]
确定所述参考基因组中多个非重叠单元中的每一个的相应单元计数参数,每个相应单元计数参数由相应单元中的dna分子位置的相应量确定;以及
[0197]
基于具有相同计数分类的连续单元进行将所述连续单元合并成合并段的分割方法,其中所述第一亚染色体区域对应于第一合并段。
[0198]
实施方案3.根据实施方案2所述的方法,其中所述分割方法包括二进制环形分割和隐马尔可夫模型中的至少一个。
[0199]
实施方案4.根据实施方案1所述的方法,其进一步包括:
[0200]
通过将其它计数参数与所述一个或多个计数阈值进行比较,检测所述生物样本中的多个其它亚染色体区域的多个畸变以确定其它计数分类;以及
[0201]
基于所述多个其它亚染色体区域的所述其它计数分类和其它尺寸分类,确定所述胎儿是否在所述多个其它亚染色体区域中具有所述多个畸变。
[0202]
实施方案5.根据实施方案1所述的方法,其中所述计数分类是过度代表或不足代表。
[0203]
实施方案6.根据实施方案1所述的方法,其中所述畸变是缺失或复制。
[0204]
实施方案7.根据实施方案1所述的方法,其中所述尺寸分类是更长、更短或相等中的一个。
[0205]
实施方案8.根据实施方案1所述的方法,其中当以下情况时,所述胎儿被确定在所述第一亚染色体区域中具有扩增:
[0206]
所述计数分类表示扩增,并且所述尺寸分类表示在所述第一亚染色体区域中不存在畸变,或
[0207]
所述计数分类表示扩增,并且所述尺寸分类表示扩增。
[0208]
实施方案9.根据实施方案8所述的方法,其进一步包括:
[0209]
当所述计数分类表示所述扩增,并且所述尺寸分类表示在所述第一亚染色体区域中不存在所述畸变时,确定所述雌性受试者在所述第一亚染色体区域中也具有所述扩增。
[0210]
实施方案10.根据实施方案8所述的方法,其进一步包括:
[0211]
当所述计数分类表示所述扩增,并且所述尺寸分类表示所述扩增时,确定所述雌性受试者在所述第一亚染色体区域中不具有所述扩增。
[0212]
实施方案11.根据实施方案1所述的方法,其中当以下情况时,所述胎儿被确定在所述第一亚染色体区域中具有缺失:
[0213]
所述计数分类表示所述缺失,并且所述尺寸分类表示在所述第一亚染色体区域中不存在所述畸变,或者
[0214]
所述计数分类表示所述缺失,并且所述尺寸分类表示所述缺失。
[0215]
实施方案12.根据实施方案11所述的方法,其进一步包括:
[0216]
当所述计数分类表示所述缺失,并且所述尺寸分类表示在所述第一亚染色体区域中不存在所述畸变时,确定所述雌性受试者在所述第一亚染色体区域中也具有所述缺失。
[0217]
实施方案13.根据实施方案11所述的方法,其进一步包括:
[0218]
当所述计数分类表示所述缺失,并且所述尺寸分类表示所述缺失时,确定所述雌性受试者在所述第一亚染色体区域中不具有所述缺失。
[0219]
实施方案14.根据实施方案1所述的方法,其中当以下情况时,所述胎儿被确定不具有所述畸变:
[0220]
所述计数分类表示扩增,并且所述尺寸分类表示缺失,或者
[0221]
所述计数分类表示所述缺失,并且所述尺寸分类表示所述扩增。
[0222]
实施方案15.根据实施方案14所述的方法,其进一步包括:
[0223]
当所述计数分类表示所述扩增,并且所述尺寸分类表示所述缺失时,确定所述雌性受试者具有所述扩增。
[0224]
实施方案16.根据实施方案14所述的方法,其进一步包括:
[0225]
当所述计数分类表示所述缺失,并且所述尺寸分类表示所述扩增时,确定所述雌性受试者具有所述缺失。
[0226]
实施方案17.根据实施方案1所述的方法,其进一步包括:
[0227]
确定所述第一亚染色体区域的第一单元的第一单元计数参数,所述第一亚染色体区域包括一个或多个单元,由位于第三区域中的dna分子的第三量和位于所述第一单元中的dna分子的第四量确定所述第一单元计数参数;
[0228]
使用对照样本计算所述第一单元的对照单元计数参数的平均值;
[0229]
通过从所述第一单元计数参数中减去对照单元计数参数的所述平均值来计算所述第一单元的分数;以及
[0230]
确定所述第一分数是否大于截止值以识别所述雌性受试者是否在所述第一亚染色体区域具有所述畸变。
foraneuploidyintoprenatalcare:whathashappenedsincetherubbermettheroad?clinchem2014;60:78
–
87.
[0256]
2.wongfck,loymd.prenataldiagnosisinnovation:genomesequencingofmaternalplasma.[epubaheadofprint]annurevmedoctober15,2015asdoi:10.1146/annurev-med-091014-115715.
[0257]
3.chiurwk,cantorcr,loymd.non-invasiveprenataldiagnosisbysinglemoleculecountingtechnologies.trendsgenet2009;25:324
–
31.
[0258]
4.petersd,chut,yatsenkosa,hendrixn,hoggewa,surtiu,etal.noninvasiveprenataldiagnosisofafetalmicrodeletionsyndrome.nengljmed2011;365:1847
–
8.
[0259]
5.jensentj,dzakulaz,deciuc,vandenboomd,ehrichm.detectionofmicrodeletion22q11.2inafetusbynext-generationsequencingofmaternalplasma.clinchem2012;58:1148
–
51.
[0260]
6.srinivasana,bianchidw,huangh,sehnertaj,ravarp.noninvasivedetectionoffetalsubchromosomeabnormalitiesviadeepsequencingofmaternalplasma.amjhumgenet2013;92:167
–
76.
[0261]
7.yuscy,jiangp,choykw,chankca,wonh-s,leungwc,etal.noninvasiveprenatalmolecularkaryotypingfrommaternalplasma.plosone2013;8:e60968.
[0262]
8.zhaoc,tynanj,ehrichm,hannumg,mcculloughr,saldivarj-s,etal.detectionoffetalsubchromosomalabnormalitiesbysequencingcirculatingcell-freednafrommaternalplasma.clinchem2015;61:608
–
16.
[0263]
9.haydenec.prenatal-screeningcompaniesexpandscopeofdnatests.nature2014;507:19
–
19.
[0264]
10.lautk,jiangfm,stevensonrj,lotk,chanlw,chanmk,etal.secondaryfindingsfromnon-invasiveprenataltestingforcommonfetalaneuploidiesbywholegenomesequencingasaclinicalservice.prenatdiagn2013;33:602
–
8.
[0265]
11.snydermw,simmonsle,kitzmanjo,coebp,hensonjm,dazarm,etal.copy-numbervariationandfalsepositiveprenatalaneuploidyscreeningresults.nengljmed2015;372:1639
–
45.
[0266]
12.yina-h,pengc-f,zhaox,caugheyba,yangj-x,liuj,etal.noninvasivedetectionoffetalsubchromosomalabnormalitiesbysemiconductorsequencingofmaternalplasmadna.procnatlacadsciusa2015;112:14670-5.
[0267]
13.yuscy,chankca,zhengywl,jiangp,liaogjw,sunh,etal.size-basedmoleculardiagnosticsusingplasmadnafornoninvasiveprenataltesting.procnatlacadsciusa2014;111:8583
–
8.
[0268]
14.chankca,zhangj,huiaby,wongn,lautk,leungtn,etal.sizedistributionsofmaternalandfetaldnainmaternalplasma.clinchem2004;50:88
–
92.
[0269]
15.loymd,chankca,sunh,chenez,jiangp,lunfmf,etal.maternalplasmadnasequencingrevealsthegenome-widegeneticandmutationalprofileofthefetus.scitranslmed2010;2:61ra91.
[0270]
16.chiurwk,chankca,gaoy,lauvym,zhengw,leungty,etal.noninvasiveprenataldiagnosisoffetalchromosomalaneuploidybymassivelyparallelgenomicsequencingofdnainmaternalplasma.procnatlacadsciusa2008;105:20458
–
63.
[0271]
17.chiurwk,akolekarr,zhengywl,leungty,sunh,chankca,etal.non-invasiveprenatalassessmentoftrisomy21bymultiplexedmaternalplasmadnasequencing:largescalevaliditystudy.bmj2011;342:c7401.
[0272]
18.palomakige,klozaemm,lambert-messerliangm,haddowje,neveuxlm,ehrichm,etal.dnasequencingofmaternalplasmatodetectdownsyndrome:aninternationalclinicalvalidationstudy.genetmed2011;13:913
–
20.
[0273]
19.hudecovai,sahotad,heungmms,jiny,leews,leungty,etal.maternalplasmafetaldnafractionsinpregnancieswithlowandhighrisksforfetalchromosomalaneuploidies.plosone2014;9:e88484.
[0274]
20.loymd,lunfmf,chankca,tsuinby,chongkc,lautk,etal.digitalpcrforthemoleculardetectionoffetalchromosomalaneuploidy.procnatlacadsciusa2007;104:13116
–
21。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1