一种再生支架型填充剂及其制备方法和应用与流程
1.本发明涉及生物医药技术领域,具体地说,是一种再生支架型填充剂及其制备方法和应用。
背景技术:
2.面部微整形美容手术,通常可通过移植自体组织或填充人工材料到皱纹区域,以达到除皱、组织提升或整体年轻化的目的。目前,市场上常用的微整形注射类填充剂有透明质酸钠类、聚乳酸类、聚己内酯类、肉毒素等。其中,聚乳酸类、聚己内酯类注射填充剂,是基于其降解产物刺激机体产生胶原蛋白,来实现面部填充。不足之处在于,此类产品有效成分的微结构皆为微球形貌,不具有理想组织再生支架材料所必需的高比表面积及高孔隙率,细胞粘附增殖效果、组织相容性仍有待提高,对于面部除皱及年轻化仍有改善空间。聚合物纤维膜通常被认为具有更高的比表面积,但聚合物纤维膜目前难以研磨至100μm以下,无法直接注射、用于美容填充剂中。因此开发新的技术方法,得到100μm以下的微纤维颗粒,有利于改善美容填充剂的效果。另外,市场上常用微整填充剂中的微球皆通过膜乳化法制备,需要经过乳化、离心、洗涤、沉淀等多步生产工艺,工艺复杂、生产周期长、成本高,故此方面也仍有需要改进的地方。
3.中国专利申请cn111298195a公开了一种复合微米材料、皮肤填充剂及其制备方法和应用,复合微米材料的制备原料包括聚氨基酸酯和脂肪族聚酯共混物、聚氨基酸~脂肪族聚酯嵌段共聚物、聚氨基酸~脂肪族聚酯接枝共聚物中的至少一种。将聚氨基酸引入脂肪族聚酯类材料,可以改善脂肪族聚酯类材料的疏水性,并且聚氨基酸在用于皮肤填充剂后,可增进皮肤弹性和保湿能力,同时可缓慢降解、不断地释放出氨基酸,能促进皮肤内新生胶原蛋白增长。另外,此复合微球制备的皮肤填充剂的细胞增殖率高于市售商品故此复合微球或微粒的皮肤填充剂可有效起到修复皱纹及凹陷的作用。但聚氨基酸的引入,使聚合物的制备过程变得复杂,增加了制剂成本及工艺生产成本。
4.而对于理想的组织工程材料来讲,其应能为细胞提供一个赖以生存的三维空间,从而有利于细胞获取足够的影响,排除代谢物,进行物质交换,使细胞能在预先涉及的支架上增殖与分化,而现有的支架要么制备方法繁琐、要么材料来源难,要么支架疗效不佳,所以在此基础上,本发明的目的一是解决高分子膜材料难以制得100μm以下的微纤维颗粒的技术困难;二是改善现有的微整填充剂因微结构皆为微球形貌而不具有理想组织再生支架材料所必需的高比表面积、高孔隙率及高细胞粘附增殖率,进而影响微整填充剂美容效果的问题;三是改善现有的微整填充剂中微球生产皆由膜乳化法制备而使得生产工艺复杂、生产周期长、生产成本高的问题。本发明通过优选相关原料以及之间的质量比和制备工艺,从而得到了一种有高比表面积、高孔隙率、高组织相容性等有优异性能的支架填充剂,关于本发明一种再生支架型填充剂及其制备方法和应用目前还未见报道。
技术实现要素:
5.本发明的目的是针对现有技术的不足,提供一种再生支架型填充剂及其制备方法和应用。
6.为实现上述目的,本发明采取的技术方案是:
7.第一方面,本发明提供了一种再生支架型填充剂,所述填充剂由以下质量比的各原料制成:聚合物微纤维颗粒:辅料=(0.1~2):(0.2~1),所述聚合物微纤维颗粒的原料是选自聚乳酸、聚己内酯、聚乙二醇、聚乙醇酸、聚对二氧环己酮和聚三亚甲基碳酸酯中的一种或两种以上的混合物,所述辅料包括悬浮剂和赋形剂中的一种或两种以上的混合物。
8.优选地,所述填充剂由以下质量比的各原料制成:聚合物微纤维颗粒:辅料=(0.5~1):(0.5~0.6)或(0.4~1.5):(0.2~0.8)。
9.优选地,所述聚合物微纤维颗粒的原料的重均分子量是(10~400)*103,所述聚合物微纤维颗粒的尺寸是10~100μm。
10.优选地,所述聚合物微纤维颗粒的原料的重均分子量是(20~200)*103或(40~300)*103或(20~30)*103或(40~60)*103或(80~100)*103或(120~150)*103或(180~200)*103。
11.优选地,所述聚合物微纤维颗粒尺寸是10~80μm或20~100μm或10~20μm或20~30μm或40~50μm。
12.优选地,所述聚合物微纤维颗粒是通过静电纺丝法或熔融纺丝法或溶液纺丝法和液氮冷冻研磨法制得。
13.优选地,所述悬浮剂选自羟丙甲基纤维素、羧甲基纤维素钠、海藻酸钠和透明质酸钠中的一种或两种以上的混合物。
14.优选地,所述赋形剂选自甘油、山梨醇、甘露醇、乙二醇、聚乙二醇和葡萄糖中的一种或两种以上的混合物。
15.优选地,所述辅料还包括溶剂、增稠剂、乳化剂中的一种或两种以上的混合物。
16.第二方面,本发明提供了如上所述的填充剂的制备方法,包括如下步骤:
17.(1)分别按照质量比称取聚合物微纤维颗粒的原料和辅料;
18.(2)将聚合物微纤维颗粒的原料按照静电纺丝法或熔融纺丝法或溶液纺丝法和液氮冷冻研磨法,制备得到聚合物微纤维颗粒;
19.(3)取步骤(1)制得的聚合物微纤维颗粒与辅料水溶液混合,所述聚合物微纤维颗粒与辅料水溶液的质量百分比为0.1%~50%,即得。
20.优选地,所述静电纺丝法制备过程包括:将包含聚乳酸、聚己内酯、聚乙二醇、聚乙醇酸、聚对二氧环己酮及聚三亚甲基碳酸酯中的一种或两种以上的混合物溶于有机溶剂中,得到纺丝原液,然后进行静电纺丝,经冷冻干燥后,得到聚合物微纤维膜;其中,纺丝原液中,制备原料的质量浓度为1~15%,优选为1.5%~10%、5%~15%,更优选为1%、5%、10%、12%。
21.其中,所述有机溶剂为二氯甲烷、氯仿、六氟异丙醇和丙酮中的一种;优选为六氟异丙醇或二氯甲烷。
22.所述静电纺丝法的工作条件为:溶液供给流量为1~20ml/h,施加电压为1~30千伏,高压端同接地端的距离为5~30厘米,作为收集装置的旋转滚筒直径为2~20厘米,旋转
滚筒的转速为50~1000转/分,纺丝时间为2~8小时。
23.优选地,所述熔融纺丝的制备过程包括:将包含聚乳酸、聚己内酯、聚乙二醇、聚乙醇酸、聚对二氧环己酮及聚三亚甲基碳酸酯中的至少一种或几种的共聚物进行真空干燥,然后进行熔融纺丝,得到聚合物纤维膜;
24.其中,所述熔融纺丝的方法可以分为直接纺丝法和切片纺丝法,优选为切片纺丝法。熔融纺丝工艺可采用高速纺丝一步法和纺丝-拉伸二步法,优选为纺丝-拉伸二步法;所述熔融纺丝的工作条件包括:真空干燥温度为40~100℃,干燥时间为10~72小时,纺丝温度为180~250℃,纺丝卷绕速度为200m/min~700m/min,喷丝板的规格为25孔*0.5mm~50孔*0.2mm,泵供量为5ml/min~20ml/min。
25.优选地,所述溶液纺丝的制备过程包括:将包含聚乳酸、聚己内酯、聚乙二醇、聚乙醇酸、聚对二氧环己酮及聚三亚甲基碳酸酯中的至少一种或几种的共聚物溶于有机溶剂中,得到纺丝原液,然后进行溶液纺丝,得到聚合物纤维膜。
26.根据本发明的制备方法,聚合物纤维膜的制备方法包括但不限于上述静电纺丝、熔融纺丝和溶液纺丝,还可以本领域其它已知方法制备。
27.优选地,所述液氮冷冻研磨法的实施过程包括:将所得的聚合物纤维膜经水溶液充分润湿,再转移到研磨罐中,研磨机频率为60hz,设定时间1分钟,经液氮冷冻,研磨成微纤维颗粒,过200-800目筛网,收集聚合物微纤维颗粒;
28.其中,所述水溶液浸润条件包括:浸润温度为5℃~-30℃,例如15℃~-25℃,示例性为25℃;浸润时间为6-72小时,例如10-36小时,示例性为12小时;研磨机的频率为40~120hz,例如50~70hz,示例性为60hz;研磨时间为0.5-5分钟,例如1-3分钟,示例性为1分钟;筛网目数为200-900目,例如230-700目,示例性为400-700目。
29.优选地,所述填充剂的形态可以为冻干粉或胶体:将所述聚合物微纤维颗粒和辅料(优选悬浮剂和赋形剂)水溶液混合冻干,得到冻干粉形态的微整填充剂;或者;
30.将所述聚合物微纤维颗粒和辅料(优选悬浮剂)水溶液混合,得到胶体形态的微整填充剂。
31.其中,所述聚合物微纤维颗粒、辅料、及其配比具有如上文所述的含义。
32.其中,所述聚合物微纤维颗粒与辅料水溶液(优选悬浮剂水溶液)的质量百分比为0.1%~50%,优选0.4%~10%、1%~15%、5%~12%,更优选为0.5%、1%、5%、8%。
33.其中,在冻干前或冻干后,可以对所述聚合物微纤维颗粒、悬浮剂和赋形剂的混合物进行分装。进一步地,所述混合、分装、冻干的过程均要求为无菌过程。
34.其中,所述聚合物微纤维颗粒和悬浮剂水溶液混合后可以进行分装。进一步地,所述混合和分装均要求为无菌过程。
35.例如,将聚合物微纤维颗粒采用冻干技术制备成冻干粉或采用预灌装针盛装得到含有聚合物微纤维颗粒的悬浮凝胶。
36.市场上常用的注射类填充剂有透明质酸钠类、聚乳酸类、聚己内酯类、肉毒素等。其中,聚乳酸类、聚己内酯类的注射填充剂,是基于刺激机体产生胶原蛋白,来实现面部填充,其形态为微球或微粒,但制作形态为纤维状网络微结构的聚合物材料用于微整填充剂的制备尚未发现。
37.采用聚合物微纤维颗粒制备的微整填充剂,解决了微球或微粒类聚合物材料的细
胞粘附问题,可以提供更好的细胞粘附支撑,使细胞在材料表面更好地粘附及生长,进而促进胶原蛋白的再生,提高填充效果及填充速度。
38.第三方面,本发明提供了如上所述的填充剂或制备方法在制备创伤组织工程修复的产品中的应用。
39.本发明优点在于:
40.1、本发明得到的再生支架型微整填充剂,其中聚合物材料具有纤维状网络微结构,此种填充剂注射皮内或皮下后,短时间内均匀分散到皱纹区域皮下组织中,之后聚合物微纤维颗粒成为包括成纤维细胞在内的各种参与细胞的再生支架,进而刺激自身胶原蛋白再生。与聚酯微球相比,聚合物微纤维颗粒由于其具有高比表面积和孔隙率,更有利于各种参与细胞的粘附及增殖,进而促进胶原蛋白的产生和合成,更好地提升面部皱纹改善及年轻化效果。此种填充剂中的悬浮剂能使聚合物微纤维颗粒均匀分散,进入皮肤后,不会发生聚合物微纤维颗粒聚集,引起肉芽肿等不良反应;赋形剂能使产品经冷冻干燥后,保持良好形貌,具有美观性。
41.2、本发明提供了所述的聚合物微纤维颗粒的制备方法,包括:通过静电纺丝或熔融纺丝或溶液纺丝技术结合超低温冷冻粉碎技术,同时在冷冻粉碎过程中,预先将纤维膜经过水溶液充分浸润,浸润进入纤维膜的水分在冷冻过程中成为冰晶,在冷冻研磨机中撞子的研磨运动中,冰晶像锋利的刀片,起到辅助切割作用,这样才能粉碎到可以直接注射的颗粒尺寸(100微米以下),制备得到尺寸及形貌可控的聚合物微纤维颗粒,解决了高分子膜材料难以制得100μm以下的微纤维颗粒的技术困难。另外,相较于微球的膜乳化法制备工艺,此制备方法简单、生产周期较短、成本较低。本发明提供的聚合物微纤维颗粒经添加悬浮剂和赋形剂后,制备成聚合物微纤维颗粒的冻干粉或悬浮凝胶,经辐照灭菌后可用于真皮深层或皮下注射进行面部皱纹与凹陷填充。本发明提供的再生支架型微整填充剂可有效起到修复面部皱纹及凹陷的作用,在医美领域具有良好的应用潜力。
附图说明
42.附图1是光学显微镜图,其中图1a是实施例1方法制得的填充剂的光学显微镜图,图1b是某市售品牌童颜针的光学显微镜图。
43.附图2是扫描电子显微镜图,其中图2a是实施例1方法制得的填充剂的扫描电子显微镜图,图2b是某市售品牌童颜针的扫描电子显微镜图。
44.附图3是各实施例的填充剂以及某市售品牌童颜针的比表面积结果。
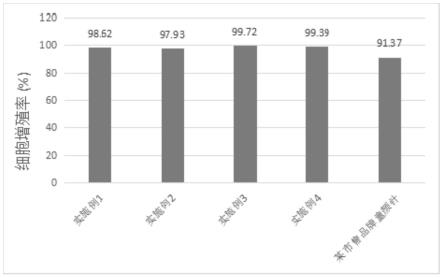
45.附图4是各实施例的填充剂以及某市售品牌童颜针的细胞增值率结果。
具体实施方式
46.下面结合具体实施方式,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明记载的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
47.除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
48.以下实施例所用的测试仪器包括:
49.光学显微镜:motic ba210数码显微镜。
50.扫描电子显微镜(sem):扫描电子显微镜(sem):zeiss gemini 300场发射扫描电镜。
51.比表面仪:asap 2020比表面积分析仪。
52.微球型微整填充剂:某市售品牌童颜针(全文所述的某市售品牌童颜针均指该产品),产品商家:吉林省杭盖秱博生物科技有限公司;产品名称:”reborn plla powder“,主要组成成分:主要由聚乳酸微球及辅料组成,所含成分不具有药理学作用,非无菌提供。
53.cck-8法测试过程:
54.(1)细胞系:小鼠胚胎成纤维细胞(nih3t3-l1,l1是通过克隆分离得到的3t3(swiss小白鼠)的连续亚株)购于中国科学院典型培养物保藏委员会细胞库。
55.(2)细胞培养:nih3t3成纤维细胞在增殖培养基(dulbecco s modified eagle培养基,dmem)中培养7天,加入10%v/v胎牛血清(fbs),37℃、95%相对湿度,5%co2。培养1天后更换培养基,去除无黏附细胞,每3天更换一次。融合时,用磷酸盐缓冲盐水(pbs)冲洗细胞两次,用胰蛋白酶/edta(0.25%w/v胰蛋白酶/0.02%edta)分离细胞5分钟,在培养基中复苏。
56.(3)细胞增殖率检测:取试验样品(实施例1-4制备得到的产品)各2个,于无菌条件下按照10mg/ml配置浸提液,每样品瓶内加入5ml浸提介质,37℃,60rmp振荡提取24小时后,取浸提液进行试验。取生长状况良好的nih3t3,消化计数,调整细胞浓度为5
×
104个/ml,在24孔板中每孔以1.25cm2/ml的浓度接种细胞,并设置阴性对照组。每组设置3个平行组。24h后向其中加入0.25mg/ml的浸提液。继续培养1天、2天、3天后,从培养箱中取出。使用cck-8检测细胞活性:将cck-8检测液与新鲜培养基以1:10的比例混合后,将24孔板内的细胞用pbs溶液清洗3次,每孔内加入110μl混合液。避光保存放入恒温箱内孵育3h后在450nm的波长下检测od值,通过空白对照组od值计算细胞相对增殖率,空白对照组记为100%。
57.复溶的方法:将实施例得到的冻干粉重新溶解于5ml灭菌注射用水中,该溶解过程主要为冻干粉中辅料的溶解,至形成聚合物微纤维颗粒的悬浮液。“悬浮时间”指的是悬浮液摇匀后到悬浮液液面开始出现分层的时间。
58.其中,聚乙醇酸、聚对二氧环己酮、聚三亚甲基碳酸酯中一种或几种的共聚物微纤维颗粒的制备过程与实施例中的聚合物微纤维颗粒制备方法相同,区别在于以聚乙醇酸、聚对二氧环己酮、聚三亚甲基碳酸酯中的一种或几种的共聚物作为原料。
59.实施例1
60.1制备方法
61.1)聚乳酸微纤维颗粒的制备
62.将重均分子量为8万的聚乳酸溶解于六氟异丙醇中,浓度为0.1g/ml,即为纺丝液。将溶液置入注射器中,注射器针头处连接高压电源。溶液供给流量控制在3ml/h,施加电压为15千伏,高压端同接地端的距离为15厘米,使用直径为8厘米的旋转滚筒作为收集装置,转速为100转/分。纺丝5小时后可收集到聚合物纤维膜。
63.将所得聚合物纤维膜经水溶液充分润湿12h,再转移到研磨罐中,研磨机频率为60hz,设定时间1分钟,经液氮冷冻,研磨成微纤维颗粒,过400-700目筛网,收集微纤维颗
粒;经asap2020比表面仪测试。
64.2)由聚乳酸微纤维颗粒制备再生支架型微整填充剂
65.将10g聚乳酸微纤维颗粒加入13g甘露醇和9g羧甲基纤维素钠的500ml水溶液中,1200转/分钟搅拌均匀后得到混悬液,每5ml混悬液灌装入西林瓶内,冻干48小时后得冻干粉,经环氧乙烷灭菌,即得到微整填充剂。其中聚乳酸微纤维颗粒、甘露醇和羧甲基纤维素钠的质量比为0.5:0.65:0.45。
66.2结果
67.聚乳酸微纤维颗粒比表面积为107m2/g,高于某市售品牌童颜针的表面积32m2/g,见图3。
68.该微整填充剂具有良好的生物相容性,经cck-8法检测其细胞增殖率为98.62%,优于某市售品牌童颜针的细胞增殖率91.37%,见图4。
69.该微整填充剂复溶后的悬浮时间为30分钟,优于聚乳酸微球微整填充剂的悬浮时间20分钟。
70.实施例2
71.1制备方法
72.1)聚己内酯微纤维颗粒的制备
73.将重均分子量为20万的聚己内酯溶解于二氯甲烷中,浓度为0.4g/ml,即为纺丝液。将溶液置入注射器中,注射器针头处连接高压电源。溶液供给流量控制在5ml/h,施加电压为15千伏,高压端同接地端的距离为18厘米,使用直径为10厘米的旋转滚筒作为收集装置,转速为120转/分。纺丝4小时后可收集到聚合物纤维膜。
74.将所得聚合物纤维膜经水溶液充分润湿10h,再转移到研磨罐中,研磨机频率为60hz,设定时间2分钟,经液氮冷冻,研磨成微纤维颗粒,过500-800目筛网,收集微纤维颗粒;经asap2020比表面仪测试。
75.2)由聚己内酯微纤维颗粒制备再生支架型微整填充剂
76.将10g聚己内酯微纤维颗粒加入含有2g交联透明质酸钠的300ml氯化钠水溶液中,混合搅拌均匀后得到复合微球的交联透明质酸钠混合凝胶,分装到预灌装注射器内,高压蒸汽灭菌,得到微整填充剂。其中聚己内酯微纤维颗粒和交联透明质酸钠的质量比为0.5:0.1。
77.2结果
78.聚己内酯微纤维颗粒比表面积为95m2/g,远高于某市售品牌童颜针的比表面积,见图3。
79.该微整填充剂具有良好的生物相容性,经cck-8法检测其细胞增殖率为97.93%,优于某市售品牌童颜针的细胞增殖率,见图4。cck-8法检测方法同实施例1。
80.实施例3
81.1制备方法
82.1)聚乳酸-聚乙二醇共聚物微纤维颗粒的制备
83.将重均分子量为18万的聚乳酸-聚乙二醇共聚物溶解于氯仿中,浓度为0.2g/ml,即为纺丝液。将溶液置入注射器中,注射器针头处连接高压电源。溶液供给流量控制在4ml/h,施加电压为18千伏,高压端同接地端的距离为15厘米,使用直径为6厘米的旋转滚筒作为
收集装置,转速为100转/分。纺丝6小时后可收集到聚合物纤维膜。
84.将所得聚合物纤维膜经水溶液充分润湿24h,再转移到研磨罐中,研磨机频率为80hz,设定时间3分钟,经液氮冷冻,研磨成微纤维颗粒,过325-600目筛网,收集微纤维颗粒;经asap2020比表面仪测试。
85.2)由聚乳酸-聚乙二醇共聚物微纤维颗粒制备再生支架型微整填充剂
86.将8g聚乳酸-聚乙二醇共聚物微纤维颗粒加入12g甘露醇和8g羧甲基纤维素钠的500ml水溶液中,1400转/分钟搅拌均匀后得到混悬液,每5ml混悬液灌装入西林瓶内,冻干48小时后得冻干粉,经环氧乙烷灭菌,即得到微整填充剂。其中聚乳酸-聚乙二醇共聚物微纤维颗粒、甘露醇和羧甲基纤维素钠的质量比为0.2:0.3:0.2。
87.2结果
88.聚乳酸-聚乙二醇共聚物微纤维颗粒比表面积为102m2/g,远高于某市售品牌童颜针的比表面积,见图3。
89.该微整填充剂具有良好的生物相容性,经cck-8法检测其细胞增殖率为99.72%,优于某市售品牌童颜针的细胞增殖率,见图4。cck-8法检测方法同实施例1。
90.该微整填充剂复溶后的悬浮时间为35分钟,优于聚乳酸微球微整填充剂的悬浮时间20分钟。
91.实施例4
92.1制备方法
93.实施例4
94.1制备方法
95.1)聚己内酯-聚乙二醇共聚物微纤维颗粒的制备
96.将重均分子量为15万的聚己内酯-聚乙二醇共聚物溶解于二氯甲烷中,浓度为0.5g/ml,即为纺丝液。将溶液置入注射器中,注射器针头处连接高压电源。溶液供给流量控制在5ml/h,施加电压为20千伏,高压端同接地端的距离为10厘米,使用直径为10厘米的旋转滚筒作为收集装置,转速为120转/分。纺丝6小时后可收集到聚合物纤维膜。
97.将所得聚合物纤维膜经水溶液充分润湿18h,再转移到研磨罐中,研磨机频率为100hz,设定时间5分钟,经液氮冷冻,研磨成微纤维颗粒,过500-800目筛网,收集微纤维颗粒;经asap2020比表面仪测试。
98.2)由聚己内酯-聚乙二醇共聚物微纤维颗粒制备再生支架型微整填充剂
99.将15g聚己内酯-聚乙二醇共聚物微纤维颗粒加入10g甘露醇和10g羧甲基纤维素钠的500ml水溶液中,1400转/分钟搅拌均匀后得到混悬液,每5ml混悬液灌装入西林瓶内,冻干48小时后得冻干粉,经环氧乙烷灭菌,即得到微整填充剂。其中聚乳酸-聚乙二醇共聚物微纤维颗粒、甘露醇和羧甲基纤维素钠的质量比为0.3:0.2:0.2。
100.2结果
101.聚己内酯-聚乙二醇共聚物微纤维颗粒比表面积为98m2/g,远高于某市售品牌童颜针的比表面积,见图3。
102.该微整填充剂具有良好的生物相容性,经cck-8法检测其细胞增殖率为99.39%,优于某市售品牌童颜针的细胞增殖率,见图4。cck-8法检测方法同实施例1。
103.该微整填充剂复溶后的悬浮时间为40分钟,优于聚己内酯微球微整填充剂的悬浮
时间30分钟。
104.实施例5
105.1实验方法
106.雄性sd大鼠,体重200-250g,共30只,均适应性喂养1周,随机分为三组,每组10只,分别为实验组、对照一组、对照二组,每组大鼠用7wt%水合氯醛腹腔注射麻醉sd大鼠,大鼠麻醉后,俯卧位固定在实验水平操作台上,剃除大鼠脊柱两侧的毛发,手术区域用碘酒消毒,然后在大鼠背部沿脊柱两侧用无菌手术刀切开一个手术切口,然后分别分离皮下组织将填充剂植入,其中实验组植入实施例1制得的填充剂,对照一组植入填充剂:组方:15g聚乳酸微纤维颗粒+1g甘露醇+0.36g羧甲基纤维素钠,对照二组植入填充剂:组方:10g聚乳酸微纤维颗粒+13g海藻糖+9g甲基丙烯酸酰化明胶。其中,对照一组、对照二组与实施例填充剂制备方法均相同,区别仅在于原料种类或各原料之间的比例不同。各组植入材料结束后,用手术线缝合大鼠背部皮肤切口,正常饲养3周,饲养3周后麻醉处死各组大鼠并取出包埋有填充剂的部分,观察并记录组织反应的程度并将组织制样做组织学评价,观察各组材料的组织相容性。
107.2结果
108.实验组填充剂呈多孔状、细丝状,内部疏松,可见较多成纤维细胞长入填充剂内部,填充剂周边可见大量纤维组织增生,淋巴细胞浸润数30-35个/hpf,中性粒细胞浸润数15-20个/hpf,巨噬细胞浸润数12-18个/hpf,多核巨细胞浸润数2-4个/hpf,毛细血管增生数2-4个/hpf。填充剂周边可见较厚的纤维囊形成,组织病理学评分为18,说明填充剂的组织相容性良好,且有新生组织长入填充剂内部。
109.对照一组填充剂呈多孔状、内部疏松但是相较于实验组,孔隙率以及疏松程度较实验组仍有一定的差异性,一些成纤维细胞长入填充剂内部,淋巴细胞浸润数23-25个/hpf,中性粒细胞浸润数10-13个/hpf,巨噬细胞浸润数6-10个/hpf,多核巨细胞浸润数1-2个/hpf,毛细血管增生数1-2个/hpf。填充剂周边可见略薄的纤维囊形成,组织病理学评分为15,说明填充剂的组织相容性较好。
110.对照二组填充剂有孔,孔隙率以及疏松程度略差,一部分成纤维细胞长入填充剂内部,淋巴细胞浸润数15-20个/hpf,中性粒细胞浸润数8-12个/hpf,巨噬细胞浸润数5-10个/hpf,多核巨细胞浸润数0-1个/hpf,毛细血管增生数0-1个/hpf。填充剂周边未见纤维囊形成,组织病理学评分为13。
111.3结论
112.上述实验结果表明,实验组较对照一组、二组的填充剂比表面积更高、孔隙率更高,组织相容性更好,成纤维细胞长入数量更多,如:淋巴细胞浸润数:对照一组、二组均是维持在25个/hpf以下,而实验组首次达到30-35个/hpf,对照一组、二组中性粒细胞浸润数均维持在13个/hpf以下,而实验组首次突破15个/hpf,高达15-20个/hpf,对照一组、二组巨噬细胞浸润数均维持在10个/hpf以下,而实验组首次突破10个/hpf,高达12-18个/hpf,以及多核巨细胞浸润数、毛细血管增生数实验组相较于对照一组、二组均有了显著性提升,而对照一组、二组与实验组区别仅在于原料种类或各原料之间的配比不同,由此可见,本发明选取的原材料种类及其之间的质量比具有协同作用。从而使得实验组较对照一组、二组的填充剂在性能上均有了质的提升。
113.以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员,在不脱离本发明原理的前提下,还可以做出若干改进和补充,这些改进和补充也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1