KrF厚膜光刻胶树脂及其制备方法和应用与流程
krf厚膜光刻胶树脂及其制备方法和应用
技术领域
[0001]
本发明涉及一种krf厚膜光刻胶树脂及其制备方法和应用。
背景技术:
[0002]
目前在半导体制造领域,lcd(液晶显示)/bump凸块/mems微机电/3d-nand存储器等芯片制造过程中,会用到krf光源厚膜光刻胶,此类光刻胶既不同于常规的krf的薄层光刻胶,也不同于arf光源的光刻胶,而是具有自己独特的性能。
[0003]
目前虽然集成电路半导体芯片制造技术在飞速发展,但配套的此类krf光源的厚膜光刻胶的技术却并没有完全成熟,是目前krf类光刻胶研究的热点领域。
[0004]
krf光源厚膜光刻胶,目前存在的问题颇多,例如,膜开裂、膜厚均匀性不佳,缺陷多,分辨率及灵敏度不佳,膜剥离性不佳,形状不佳,矩形性差,解析性差,耐热性不够强、并且不能抑制波动现象,以及杂质严重等缺陷。
[0005]
因此,本领域亟需开发能综合解决上述问题的厚膜光刻胶。
技术实现要素:
[0006]
本发明所要解决的技术问题是现有技术中krf光源厚膜光刻胶膜开裂、膜厚均匀性不佳、缺陷多、分辨率及灵敏度不佳、膜剥离性不佳、形状不佳、矩形性差、解析性差、耐热性不够强、不能抑制波动现象或者杂质严重等缺陷,为此,提供了一种krf厚膜光刻胶树脂及其制备方法和应用。本发明的光刻胶组合物形成的胶膜至少具有如下任一优点:不易开裂、厚度均匀、分辨率及灵敏度不佳、膜剥离性佳、形状佳、矩形性佳、解析性佳、耐热性好、还能抑制波动现象以及金属杂质少。
[0007]
本发明通过以下技术方案来解决上述技术问题。
[0008]
本发明提供了一种光刻胶组合物,其包括以下组分:光致产酸剂、树脂和溶剂;
[0009]
所述的光致产酸剂为pag1和/或pag2,其结构如下所示:
[0010][0011]
所述的树脂通过以下制备方法制得,所述的树脂的制备方法包括以下步骤:
[0012]
在过氧苯甲酰存在下,将如式a所示的单体、如式b所示的单体、如式c所示的单体、如式d所示的单体和如式e所示的单体在乙酸乙酯中进行聚合反应,得到所述的树脂;其中,所述的如式d所示的单体以重量份数计的重量份数为1-10份,所述的如式e所示的单体以重
量份数计的重量份数为1-10份;
[0013]
所述的聚合反应的温度为75-80℃;
[0014][0015]
式a中,r1为r
1a
取代的5-10元的杂环烷基或-ch2(c=o)or
1b
;
[0016]
r
1b
为r
1b-1
取代的5-10元的杂环烷基;
[0017]
r
1a
和r
1b-1
独立地为氧代、氰基或c
1-4
的烷基;
[0018]
所述的r
1a
取代的5-10元的杂环烷和所述的r
1b-1
取代的5-10元的杂环烷基中的杂原子为o,个数为1个或2个;
[0019]
式b中,r2为
[0020]
n1为1-11中的任意整数;
[0021]
r
2a
和r
2b
独立地为c
1-4
的烷基、羟基取代的c
1-4
的烷基、苯基、r
2a-1
取代的苯基、5-6元的环烷基或金刚烷基;
[0022]
r
2a-1
为c
1-4
的烷基或c
1-4
的烷氧基;
[0023]
或者,r
2a
和r
2b
与其相连的碳原子一起形成5-6元的杂环烷基或r
2b-1
取代的5-6元的杂环烷基,所述的5-6元的杂环烷基和r
2b-1
取代的5-6元的杂环烷基中的杂原子独立地选自o和n,个数为1个或2个;
[0024]
r
2b-1
为c
1-4
的烷基或氨基保护基;
[0025]
式c中,为单键或者双键;
[0026]
r
3a
、r
3b
和r
3c
独立地为h、羟基、氰基、-(c=o)or
3a-1
、-o(c=o)r
3a-2
、c
1-4
的烷基或羟基取代的c
1-4
的烷基;且r
3a
、r
3b
和r
3c
不同时为h;
[0027]
r
3a-1
为h、c
1-5
的烷基、
[0028]
r
3a-2
为c
1-4
的烷基或苯基;
[0029]
或者,r
3a
、r
3b
和r
3c
中的任意两个基团与其相连的碳原子一起形成苯基、5-7元的环烷基、5-7元的环烯基、
[0030]
式d中,n2为0或1;
[0031]
r
4a
和r
4b
独立地为h或c
1-4
的烷基;且r
4a
和r
4b
不同时为h;
[0032]
式e中,r5为h、氰基、c
1-4
的烷基、r
5a
取代的c
1-4
的烷基或-(c=o)or
5b
;
10元的杂环烷基优选为10元的杂环烷基优选为更优选为
[0049]
本发明中,当r
2a
和r
2b
独立地为c
1-4
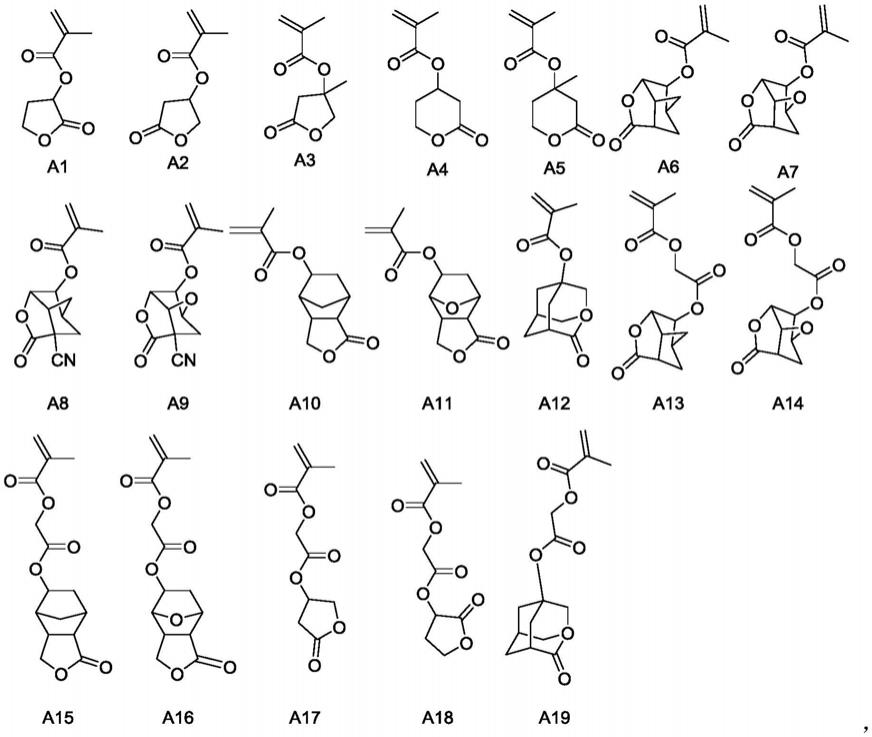
的烷基时,所述的c
1-4
的烷基优选为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基或叔丁基。
[0050]
本发明中,当r
2a-1
为c
1-4
的烷基时,所述的c
1-4
的烷基优选为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基或叔丁基,更优选为甲基。
[0051]
本发明中,当r
2a
和r
2b
独立地为羟基取代的c
1-4
的烷基时,所述的c
1-4
的烷基优选为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基或叔丁基,更优选为乙基。
[0052]
本发明中,当r
2a-1
为c
1-4
的烷氧基时,所述的c
1-4
的烷氧基优选为甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、仲丁氧基、异丁氧基或叔丁氧基,更优选为甲氧基或正丁氧基、仲丁氧基、异丁氧基或叔丁氧基。
[0053]
本发明中,当r
2a
和r
2b
独立地为5-6元的环烷基时,所述的5-6元的环烷基优选为环戊基或环己基,更优选环己基。
[0054]
本发明中,当r
2a
和r
2b
与其相连的碳原子一起形成5-10元的杂环烷基时,所述的5-6元的杂环烷基优选为
[0055]
本发明中,当r
2a
和r
2b
与其相连的碳原子一起形成r
2b-1
取代的5-6元的杂环烷基时,所述的5-6元的杂环烷基优选为6杂环烷基。
[0056]
本发明中,当r
2a
和r
2b
与其相连的碳原子一起形成r
2b-1
取代的5-6元的杂环烷基时,所述的5-6元的杂环烷基中的杂原子优选为n。
[0057]
本发明中,当r
2b-1
为c
1-4
的烷基时,所述的c
1-4
的烷基优选为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基或叔丁基,更优选为甲基、正丁基、仲丁基、异丁基或叔丁基。
[0058]
本发明中,当r
2a
和r
2b
与其相连的碳原子一起形成r
2b-1
取代的5-6元的杂环烷基时,所述的r
2b-1
取代的5-6元的杂环烷基优选为
[0059]
本发明中,当r
3a
、r
3b
和r
3c
独立地为c
1-4
的烷基时,所述的c
1-4
的烷基优选为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基或叔丁基,更优选为叔丁基。
[0060]
本发明中,当r
3a
、r
3b
和r
3c
独立地为羟基取代的c
1-4
的烷基时,所述的c
1-4
的烷基优
选为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基或叔丁基,更优选为甲基。
[0061]
本发明中,当r
3a-1
为c
1-5
的烷基时,所述的c
1-5
的烷基优选为乙基或
[0062]
本发明中,当r
3a-1
为c
6-10
的芳基时,所述的c
6-10
的芳基优选为苯基。
[0063]
本发明中,当r
3a-2
为c
1-4
的烷基时,所述的c
1-4
的烷基优选为甲基。
[0064]
本发明中,当r
3a-2
为c
6-10
的芳基时,所述的c
6-10
的芳基优选为苯基。
[0065]
本发明中,当r
3a
、r
3b
和r
3c
中的任意两个基团与其相连形成5-7元的环烷基时,所述的5-7元的环烷基优选为环戊基、环己基或
[0066]
本发明中,当r
3a
、r
3b
和r
3c
中的任意两个基团与其相连形成5-7元的环烯基时,所述的5-7元的环烯基优选为环戊烯基。
[0067]
本发明中,当r
4a
和r
4b
独立地为h或c
1-4
的烷基时,所述的c
1-4
的烷基优选为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基或叔丁基,更优选为甲基、乙基或正丙基。
[0068]
本发明中,当r5为r
5a
取代的c
1-4
的烷基时,所述的c
1-4
的烷基优选为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基或叔丁基,更优选为叔丁基。
[0069]
本发明中,当r
5b
为c
1-4
的烷基时,所述的c
1-4
的烷基优选为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基或叔丁基,更优选为甲基或乙基。
[0070]
本发明中,当r
5b
为c
1-4
的烷基时,所述的c
1-4
的烷基优选为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基或叔丁基,更优选为甲基。
[0071]
本发明中,所述的如式a所示的单体优选为如下任一化合物:
[0072][0073]
更优选为如下任一化合物:
[0074][0075]
本发明中,所述的如式b所示的单体优选如下任一化合物:
[0076]
[0077][0078]
更优选如下任一化合物:
[0079][0080]
本发明中,所述的如式c所示的单体优选为如下任一化合物:
[0081]
更优选为如下任一化合物:
[0082][0083]
本发明中,所述的如式d所示的单体优选为如下任一化合物:
[0084]
本发明中,所述的如式e所示的单体优选为如下任一化合物:
[0085][0086]
更优选为如下任一化合物:
[0087][0088]
本发明中,所述的树脂通过以下制备方法制得,所述的树脂的制备方法中,各单体及用量为如下(1)-(10)任一组所示,相应地依次得到树脂1至树脂10:
[0089]
(1)30份所述的如式a所示的单体、40份所述的如式b所示的单体、20份所述的如式c所示的单体、5份所述的如式d所示的单体和5份所述的如式e所示的单体;
[0090]
所述的如式a所示的单体为所述的如式b所示的单体为所述的如式c所示的单体为所述的如式d所示的单体为
所述的如式e所示的单体为以此得到树脂1;
[0091]
(2)35份所述的如式a所示的单体、45份所述的如式b所示的单体、10份所述的如式c所示的单体、5份所述的如式d所示的单体和5份所述的如式e所示的单体;
[0092]
所述的如式a所示的单体为所述的如式b所示的单体为所述的如式c所示的单体为所述的如式d所示的单体为所述的如式e所示的单体为以此得到树脂2;
[0093]
(3)40份所述的如式a所示的单体、48份所述的如式b所示的单体、10份所述的如式c所示的单体、1份所述的如式d所示的单体和1份所述的如式e所示的单体;
[0094]
所述的如式a所示的单体为所述的如式b所示的单体为所述的如式c所示的单体为所述的如式d所示的单体为所述的如式e所示的单体为以此得到树脂3;
[0095]
(4)30份所述的如式a所示的单体、40份所述的如式b所示的单体、15份所述的如式c所示的单体、5份所述的如式d所示的单体和10份所述的如式e所示的单体;
[0096]
所述的如式a所示的单体为所述的如式b所示的单体为所述的如式c所示的单体为所述的如式d所示
的单体为所述的如式e所示的单体为以此得到树脂4;
[0097]
(5)40份所述的如式a所示的单体、35份所述的如式b所示的单体、10份所述的如式c所示的单体、10份所述的如式d所示的单体和5份所述的如式e所示的单体;
[0098]
所述的如式a所示的单体为所述的如式b所示的单体为所述的如式c所示的单体为所述的如式d所示的单体为所述的如式e所示的单体为以此得到树脂5;
[0099]
(6)30份所述的如式a所示的单体、40份所述的如式b所示的单体、20份所述的如式c所示的单体、5份所述的如式d所示的单体和5份所述的如式e所示的单体;
[0100]
所述的如式a所示的单体为所述的如式b所示的单体为所述的如式c所示的单体为所述的如式d所示的单体为所述的如式e所示的单体为以此得到树脂6;
[0101]
(7)35份所述的如式a所示的单体、45份所述的如式b所示的单体、10份所述的如式c所示的单体、5份所述的如式d所示的单体和5份所述的如式e所示的单体;
[0102]
所述的如式a所示的单体为所述的如式b所示的单体为
所述的如式c所示的单体为所述的如式d所示的单体为所述的如式e所示的单体为以此得到树脂7;
[0103]
(8)40份所述的如式a所示的单体、40份所述的如式b所示的单体、10份所述的如式c所示的单体、5份所述的如式d所示的单体和5份所述的如式e所示的单体;
[0104]
所述的如式a所示的单体为所述的如式b所示的单体为所述的如式c所示的单体为所述的如式d所示的单体为所述的如式e所示的单体为以此得到树脂8;
[0105]
(9)30份所述的如式a所示的单体、40份所述的如式b所示的单体、15份所述的如式c所示的单体、5份所述的如式d所示的单体和10份所述的如式e所示的单体;
[0106]
所述的如式a所示的单体为所述的如式b所示的单体为所述的如式c所示的单体为所述的如式d所示的单体为所述的如式e所示的单体为以此得到树脂9;
[0107]
(10)40份所述的如式a所示的单体、35份所述的如式b所示的单体、10份所述的如式c所示的单体、10份所述的如式d所示的单体和5份所述的如式e所示的单体;
[0108]
所述的如式a所示的单体为所述的如式b所示的单体为所述的如式c所示的单体为所述的如式d所示的单体为所述的如式e所示的单体为以此得到树脂10。
[0109]
所述的聚合反应中,所述的过氧苯甲酰与“如式a所示的单体、如式b所示的单体、如式c所示的单体、如式d所示的单体和如式e所示的单体”的质量比可以为本领域常规的质量比,例如1:50。
[0110]
所述的聚合反应中,所述的聚合反应的温度优选77℃。
[0111]
所述的聚合反应中,所述的乙酸乙酯与所述的“如式a所示的单体、如式b所示的单体、如式c所示的单体、如式d所示的单体和如式e所示的单体”的质量比可以为本领域常规的质量比,例如6:5。
[0112]
所述的聚合反应中,所述的聚合反应的时间可以为本领域常规的时间,例如7小时。
[0113]
所述的聚合反应结束后的后处理优选如下步骤:冷却、沉淀和干燥。
[0114]
其中,所述的沉淀的次数可以本领域的常规沉淀次数,例如3次。
[0115]
其中,所述的沉淀中所用的溶剂可以为醇类溶剂,进一步可以为甲醇。
[0116]
其中,所述的干燥优选在真空干燥箱中干燥。
[0117]
在本发明一优选实施方式中,所述的树脂通过具体实施方式中的树脂的制备中的方法制得(即树脂的制备中的步骤1-3)。
[0118]
本发明中,所述的溶剂以重量份数计的份数可以为本领域常规的份数,优选50-85份,例如59-70份,再例如60份、69.4份、77份。
[0119]
本发明中,所述的溶剂可以为本领域常规的种溶剂,优选酮类溶剂、酯类溶剂和醚类溶剂中的一种或多种。当所述的溶剂为酮类溶剂时,所述的酮类溶剂可以为环己酮。所述的酯类溶剂可以为乙酸乙酯。所述的醚类溶剂可以为乙二醇单甲醚和/或二丙二醇单甲醚。
[0120]
在本发明一优选实施方式中,所述的光刻胶组合物还含有添加剂,所述添加剂选自流平剂、增塑剂、有机碱、溶解速度增强剂和光敏剂中的至少一种。所述的添加剂以重量份数计的份数优选为5-10份。
[0121]
在本发明一优选实施方式中,所述的树脂的制备方法包括以下步骤:
[0122]
氮气保护下,将“乙酸乙酯和过氧苯甲酰”的混合液加入到“如式a所示的单体、如式b所示的单体、如式c所示的单体、如式d所示的单体、如式e所示的单体和乙酸乙酯”的混合溶液中;
[0123]
其中,所述的加入的时间优选为10min。
[0124]
在本发明一优选实施方式中,所述的光刻胶组合物由以下组分组成:所述的光致产酸剂、所述的树脂和所述的溶剂;所述的光致产酸剂的份数、所述的树脂的种类和份数、“所述的溶剂的种类和份数”均同前所述。
[0125]
在本发明一优选实施方式中,所述的光刻胶组合物由以下组分组成:所述的光致产酸剂、所述的树脂、所述的溶剂和所述的添加剂;所述的光致产酸剂的份数、所述的树脂的种类和份数、所述的溶剂的种类和份数、和“所述的添加剂的种类和份数”均同前所述。
[0126]
在本发明一优选实施方式中,所述的光刻胶组合物为下述任一组合:
[0127]
组合1:5份的pag1,25份的树脂1和70份的环己酮;
[0128]
组合2:0.6份的pag2,30份的树脂2和69.40份的乙酸乙酯;
[0129]
组合3:5份的pag2,10份的树脂3和85份的乙二醇单甲醚;
[0130]
组合4:5份的pag1,35份的树脂4和60份的环己酮;
[0131]
组合5:5份的pag2,45份的树脂5和50份的乙二醇单甲醚;
[0132]
组合6:5份的pag1,25份的树脂6和70份的乙酸乙酯;
[0133]
组合7:0.6份的pag2,30份树脂7和69.4份的二丙二醇单甲醚;
[0134]
组合8:3份的pag1,20份树脂8和77份的二丙二醇单甲醚;
[0135]
组合9:5份的pag2,35份的树脂9和60份的乙酸乙酯;
[0136]
组合10:5份的pag2,45份的树脂10和50份的乙二醇单甲醚;
[0137]
其中,所述的树脂1、所述的树脂2、所述的树脂3、所述的树脂4、所述的树脂5、所述的树脂6、所述的树脂7、所述的树脂8、所述的树脂9和所述的树脂10均如前所述。
[0138]
本发明还提供了一种光刻胶组合物的制备方法,其包括以下步骤:将上述各组分混合均匀,即可。
[0139]
所述的制备方法中,所述的混合的方式可以为本领域常规的混合方式,优选震荡。
[0140]
所述的制备方法中,所述的混合的时间可以为本领域常规的混合时间,优选18-30小时,24小时。
[0141]
所述的混合后,还可进一步包括过滤步骤。所述的过滤的方式可以为本领常规的方式,优选采用过滤器过滤。所述的过滤的次数优选2-3次,例如2次。所述的过滤器的滤膜孔径优选为20-50nm或2-20nm。当所述的过滤的次数为2次时,第一过滤器的滤膜孔径大于第二过滤器的滤膜孔径。所述的第一过滤器的滤膜孔径优选为20-50nm。所述的第一过滤器的滤膜孔径优选为2-20nm。
[0142]
本发明还提供了一种形成光刻图案的方法,所述的方法包括如下步骤:
[0143]
步骤1:将上述的的光刻胶组合物涂覆在基材表面以形成光刻胶组合物层;
[0144]
步骤2:对所述的光刻胶组合物层进行烘烤;
[0145]
步骤3:将烘烤后的光刻胶组合物层进行冷却;
[0146]
步骤4:通过曝光将掩膜板上的图案复制到烘烤后的光刻胶组合物层上;
[0147]
步骤5:将曝光后的光刻胶组合物层进行烘烤;
[0148]
步骤6:向烘烤后的光刻胶组合物层施加显影剂进行显影,即可得到光刻图案。
[0149]
步骤1中,所述的基材优选采用hmds进行预处理。
[0150]
步骤1中,所述的基材优选为12片硅片。
[0151]
步骤1中,所述的涂覆的方式优选旋涂。
[0152]
步骤1中,所述的光刻胶组合物层的厚度优选为2.5-5μm(例如2.5μm、3.6μm、4.5μm、5.0μm)。
[0153]
步骤2中,所述的烘烤的温度优选为120-150℃。
[0154]
步骤2中所述的烘烤的时间优选为80-150秒。
[0155]
步骤3中,所述的冷却的温度优选为冷却至室温。
[0156]
步骤4中,所述的曝光的波长优选为248nm。
[0157]
步骤4中,所述的曝光的强度优选为10-50mj/cm2。
[0158]
步骤5中,所述的烘烤的温度优选为90-120℃。
[0159]
步骤5中,所述的烘烤的时间优选为90-130秒。
[0160]
步骤6中,所述的显影剂优选为四甲基氢氧化铵(tmah)水溶液,例如2.38%tmah。
[0161]
步骤6中,所述的显影的时间优选为50-70秒,例如60秒。步骤6中显影结束后,还可包括冲洗步骤所述的冲洗采用的溶剂为水例如纯水
[0162]
本发明中,“烷基”是指具有指定的碳原子数的直链或支链烷基。
[0163]
本发明中,“环烷基”是指具有稳定的环状体系的饱和的单环环体系(例如环戊基、环己基)或者饱和的2-4元桥环体系(例如)。
[0164]
本发明中,“杂环烷基”,指含1个或多个n、o或s的杂原子的单环(例如,指含1个或多个n、o或s的杂原子的单环(例如)或2-4元桥环体系(例如)。
[0165]
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0166]
本发明的树脂为自制,其他所用试剂和原料均市售可得。
[0167]
本发明的积极进步效果在于:采用本发明的光刻胶组合物形成的胶膜至少具有如下任一优点:不易开裂、厚度均匀、分辨率及灵敏度不佳、膜剥离性佳、形状佳、矩形性佳、解析性佳、耐热性好、还能抑制波动现象以及金属杂质少。
具体实施方式
[0168]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0169]
下述实施例及对比例中,未限定具体操作温度的,均是在室温条件下进行。
[0170]
树脂的制备
[0171]
实施例或者对比例中所用到的树脂1-15按照下述方法制得。所用的各个单体如下
所示:
[0172]
单体a:
[0173][0174]
单体b:
[0175]
单体c:
[0176]
单体d:
[0177][0178]
单体e:
[0179][0180]
步骤1:将按照表1中a-e的单体的量,加入到充满氮气的反应釜内,然后在反应釜内加入100g乙酸乙酯,搅拌均匀后将反应釜升温至77℃,接着再次向反应釜中滴加乙酸乙酯(20g)和过氧苯甲酰(2g)的混合液,10min滴加完毕。在77℃下反应7小时,停止反应,将反应液的温度冷却至室温;
[0181]
步骤2:反应液冷却至室温后,向反应釜中加入甲醇(1000g),产生沉淀。1h后,导出出反应釜内的液体,向反应釜内加入乙酸乙酯至沉淀溶解(120g);
[0182]
步骤3:向步骤2中的反应釜内加入甲醇(1000g),重复步骤2的操作3次,得到固体沉淀物。将固体沉淀物置于真空干燥箱中干燥,得到78g改性成膜树脂。
[0183]
采用凝胶渗透色谱(gpc)设备测量改性成膜树脂的重均分子量及聚合物分散性指数(pdi)。
[0184]
表1
[0185]
[0186][0187]
下述实施例或对比例中,光刻胶组合物均按照下述方法制备:
[0188]
实施例1-10及对比例1-10
[0189]
光致产酸剂:
[0190][0191]
按照表2中原料的加入到一个新的干净的100ml玻璃瓶中。室温下,混合物在瓶中震荡24小时,使其充分溶解,然后先后用0.22微米和0.02微米的过滤器过滤光刻胶溶液,得到光刻胶组合物。
[0192]
表2
[0193]
实施例编号树脂序号光致产酸剂溶剂树脂重量pag重量溶剂重量11pag1环己酮25g5g70g22pag2乙酸乙酯30g0.60g69.40g33pag2乙二醇单甲醚10g5g85g44pag1环己酮35g5g60g55pag2乙二醇单甲醚45g5g50g66pag1乙酸乙酯25g5g70g77pag2二丙二醇单甲醚30g0.60g69.40g88pag1二丙二醇单甲醚20g3g77g99pag2乙酸乙酯35g5g60g1010pag2乙二醇单甲醚45g5g50g对比例111pag1环己酮25g5g70g对比例212pag1环己酮25g5g70g对比例313pag1环己酮25g5g70g对比例414pag1环己酮25g5g70g对比例515pag1环己酮25g5g70g对比例61pag3环己酮25g5g70g对比例71pag4环己酮25g5g70g对比例82pag3乙酸乙酯30g0.60g69.40g对比例92pag4乙酸乙酯30g0.60g69.40g对比例212pag4环己酮25g5g70g
[0194]
效果实施例
[0195]
在匀胶机的hmds腔体中,将气态hmds沉积到晶圆衬底表面。将实施例1-10以及对比例1-10的光刻胶旋涂在hmds预处理的12”硅片上,以1000~3000转/分钟的速度旋转成膜,120℃热板上烘烤90秒,冷板腔体中冷却至室温,然后在曝光机上曝光,曝光机的波长为248nm,曝光强度10-50mj/cm2。曝光后在110℃热板上烘烤90秒,最后在2.38%tmah显影液中显影60秒,采用纯水进行冲洗,然后烘干在电子显微镜检测光刻结果。
[0196]
光刻机型号为248nm krfstepper:nikon s204b,0.55na,0.33sigma(na:numerical aperture数值孔径;sigma:光圈)。
[0197]
1、抗裂性
[0198]
采用sem设备(设备名“s8840”;hitachi corporation制造)观察光致抗蚀剂膜表面抗裂性。
[0199]
2、黏度测试
[0200]
使用自动黏度测定装置vmc-252(离合公司制)测定厚膜光刻胶的黏度(25℃)。
[0201]
3、膜厚测量
[0202]
用光刻胶膜厚测定装置纳米测试器(nanometrics公司制)对晶圆49处测定点的光刻胶膜厚(nm)进行测定。
[0203]
4、膜厚面内均匀性评价
[0204]
3σ为49处测定结果所算出的标准误差(σ)的3倍值(3σ)。该3σ之数值越小时,其表示膜厚的面内不均匀度越小,而可得到具有高质量面内均匀性的光刻胶。
[0205]
5、缺陷评价
[0206]
缺陷通过由kla-tencorcorporation制造的表面缺陷测定装置kla2132(商品名)测定,评价在基材上的缺陷数目。
[0207]
6、分辨限度cd(critical dimension)(nm))
[0208]
利用电子显微镜进行观察,将抗蚀剂图案中的非抗蚀剂部(线/空间=4:1)的截面中的、基板界面的空间宽度作为分辨限度cd进行了评价。
[0209]
7、形状评价
[0210]
由显影后的晶片的截面sem的结果,能够分辨至基板,将图案的直线性良好者(矩形状者)评价为a,将无法分辨至基板但图案的直线性差者(底部突出者)评价为b。
[0211]
8、图案的截面形状的矩形性评价
[0212]
借由sem观察图案的截面形状,将侧面几乎垂直地切除者设为a,将成为大致圆锥状者设为b,将侧面成为波状者设为c,将侧面成为波状者设为d。
[0213]
9、解析性的评价
[0214]
使用遮罩,使曝光量发生变化,观察了光刻胶图案的空间宽度。将空间宽度中最微细的空间宽度(单位:微米)作为解析性评价的指标。
[0215]
10、真空处理后的膜剥离的评价
[0216]
将评价用图案晶圆放入耐压容器中,并进行了真空处理(以0.01torr放置了15分钟)。利用光学显微镜(olympus corporation制)在光学显微镜模式下使用扫描型共聚焦雷射显微镜(型号:lext ols3100)观察真空处理后的晶圆,观察了晶圆表面的膜剥离。将膜剥离超过100处的多数情况计数为d、膜剥离为6~100处的情况计数为c、膜剥离为2~5处的情况计数为b、膜剥离为1处的情况计数为a、膜剥离为0处的情况计数为s。
[0217]
11、灵敏度评价
[0218]
按照前述光刻方法形成由每个宽度为1.5μm的直线-和-间隔宽度(l&s)(1∶1)组分的图案所需的曝光时间用毫秒(ms)单位(eop曝光量)表示。
[0219]
12、波动现象的评价
[0220]
使用临界尺寸测量sem,从右侧穿过具有1.5μm宽度的l&s抗蚀剂图案观察所获得抗蚀剂图案的外形。不能识别波动现象的情况被定为“a”,而能识别波动现象的情况被定为“b”。
[0221]
13、景深(dof)的测量
[0222]
掩膜图案的规定尺寸(线宽:1.5μm,l&s抗蚀剂图案:1∶1)所需的eop曝光量用作标准曝光量,然后,l&s(线宽:1.5μm,l&s抗蚀剂图案:1∶1)外形的横截面sem照片使用sem在上下移动焦点的条件下的照射剂量中拍摄,然后曝光并观察进一步的显影。为在sem显微照片中的
±
10%的规定尺寸内获得具有1.5μm宽度的矩形抗蚀剂图案所需要的焦点偏离的最大值(μm)被认为是景深。
[0223]
14、耐热性测试
[0224]
在上述的相同方式中,具有1.5μm宽度的l&s抗蚀剂图案在通过上述灵敏度测试获得的曝光量(eop)中形成,并进行在140℃的热处理300秒。然后,通过sem观察横截面外形。抗蚀剂图案的形变几乎不能观察到的情况被定为“a”,而可观察到抗蚀剂图案收缩的情况被定为“b”。
[0225]
15、金属杂质
[0226]
用agilent technologies公司制icp-ms装置(电感耦合等离子体质谱仪)“agilent7500cs”测量了各组成物中所含的25种(na、k、ca、fe、cu、mg、mn、al、li、cr、ni、sn、zn、ag、as、au、ba、cd、co、pb、ti、v、w、mo、zr)的金属杂质成分量,将含量最高的金属杂质的含量大于10ppb记为b;小于10ppb记为a。
[0227]
表3
[0228][0229][0230]
表4
[0231][0232]
备注:表3和4中的“/”表示没有进行测试。
[0233]
由上表3可知,本发明的光刻胶组合物形成的厚膜无开裂,膜厚均匀性较好(3σ在42以下)、缺陷少,分辨率在500-1000nm,横截面形状均为矩形,解析性好(抗蚀剂图案的空间宽度在1.1-1.8微米),处理后的膜剥离少好(sabc),灵敏度高(250-300ms),耐热性强(抗蚀剂图案的形变几乎不能观察到的情况)、能抑制波动现象,以及杂质少(最少可以达到小于10ppb)。
[0234]
以真空处理后的膜剥离的评价为例,通过表3和4中的“实施例1和对比例1-5”(改变树脂的种类)、“实施例1和对比例6-7”和“实施例2和对比例8-9”(改变光致产酸剂种类的)、“实施例1和对比例10”(同时改变树脂和光致产酸剂的种类)的真空处理后的膜剥离的评价指标对比可知:本发明的光刻胶组合物形成的胶膜的膜剥离处均较不在本发明范围内的树脂、光致产酸剂范围内、或者“树脂和光致产酸剂”的光刻胶组合物形成的胶膜的膜剥离处少。本发明范围内的光刻胶组合物形成的胶膜膜剥离处较少。可见本发明范围内的光
刻胶组合物具有较好的性能。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1