橡胶增强填充用水合硅酸的制作方法
本发明涉及橡胶增强填充用水合硅酸。详细而言,本发明提供一种橡胶增强填充用水合硅酸,其在混配到轮胎或各种工业用橡胶产品中时能够实现高度的分散性,并且可获得高增强性(特别是耐磨耗性)。
关联申请的相互参照
本申请要求2018年8月10日提交的日本专利申请2018-151033号的优先权,其全部记载特别地作为公开内容援引于本说明书中。
背景技术:
作为橡胶的增强填充剂,一直以来使用了炭黑。另一方面,近年来,出于能够降低滚动阻力、并且易于进行橡胶的彩色化等原因,开始广泛使用水合硅酸作为白色系的增强填充剂。特别是在将混配有水合硅酸的橡胶用于轮胎胎面的情况下,由于能够降低滚动阻力,因而近年来混配有水合硅酸的充气轮胎的胎面受到关注。此外,出于对环境的考虑,还需要改善橡胶的增强性(特别是耐磨耗性)的水合硅酸,对于水合硅酸的要求越来越高。
水合硅酸在表面具有硅烷醇基(-sioh),其与橡胶分子结合而有助于增强性的提高。比表面积越高(后述的一次颗粒越小),水合硅酸的硅烷醇基个数越增加。因此,据称具有越高比表面积的水合硅酸越能提高橡胶的增强性。
作为水合硅酸使橡胶增强性提高的主要原因,可以举出下述两点:(1)bet比表面积高;以及(2)分散性良好。水合硅酸具有10~60nm左右的一次颗粒聚集成2~100μm左右的粒径而成的结构,bet比表面积约为50~250m2/g。还已知:bet比表面积越高则一次颗粒越小,表面的硅烷醇基彼此通过氢键密集地聚集,内聚力增强。另一方面,在橡胶增强填充用水合硅酸的情况下,提高比表面积也很重要,但同时分散性越高则橡胶增强性越增加。从该方面出发,优选内聚力弱。
作为着眼于bet比表面积的技术,已知有通过进一步提高bet比表面积、进而加入添加剂来提高橡胶增强性的尝试(专利文献1、2)
专利文献1公开了一种橡胶用水合硅酸的制法。通过加入多元羧酸、铝,提高了聚合物与水合硅酸的结合力,从而提高了增强性。但是,专利文献1所记载的制法中使用酸性反应,因此所得到的水合硅酸的内聚力增强,分散性存在困难。
专利文献2公开了一种涉及在表面附加了铝的水合硅酸的发明。对于比表面积高(此处ctab比表面积为160m2/g以上)且具有特定的氮气孔分布的水合硅酸,橡胶分子容易侵入。提出了通过在这种水合硅酸的表面附加铝而提高与橡胶分子的结合力、从而提高增强性的方案。
关于水合硅酸的分散性改善,在专利文献3中有公开。专利文献3以wk率这种尺度来评价分散性,提出了wk率低、分散性良好的水合硅酸。专利文献3中,在提高分散性的同时,通过向水合硅酸中加入al2o3来提高聚合物与水合硅酸的结合力,还实现了增强性的提高。专利文献3中记载的水合硅酸(沉降硅酸)具有2~5.0重量%的al2o3含量和小于3.4的wk率、80~139m2/g的ctab表面积、80~180m2/g的bet比表面积。
专利文献1:日本特表2017-514773号公报(wo2015/121332)
专利文献2:日本特开2017-2210号公报
专利文献3:日本特开2000-72434号公报
专利文献1~3的全部记载特别地作为公开内容援引于本说明书中。
技术实现要素:
发明所要解决的课题
与以往相比更需要如下所述的水合硅酸:进一步提高增强性、特别是耐磨耗性是当然的,而且在橡胶中的分散性良好,混炼作业时的作业性良好。但是,专利文献1、2中记载的水合硅酸虽然提高了比表面积,但如上所述高比表面积的水合硅酸由于一次颗粒小的原因,聚集结构增强,存在无法得到充分的分散性的问题。专利文献3中记载的水合硅酸虽然能够在某种程度上改善分散性,但比表面积不充分,增强性不充分。
另外,在为了提高处理性能而将比表面积高的水合硅酸制成粒状的情况下,聚集结构(与比表面积低的情况相比)变得更强,存在导致分散性进一步恶化的问题。特别是在通过酸性反应进行中和合成的专利文献1的方法中,具有能够容易地提高bet比表面积的优点,同时在将水合硅酸混配(混炼)到橡胶中时,在初始阶段水合硅酸被较早地捏合,因此处理性能非常优异。但是,另一方面也具有下述缺点:即使在橡胶混配的最后阶段,水合硅酸在橡胶中也不分散,无法得到充分的增强性。这些分散不良不仅不会有助于增强性能的提高,而且从品质稳定性的问题来看,可以说也是越少越好。
这样,需要开发出一种水合硅酸,其为具有比较高的bet比表面积(例如230m2/g以上)的橡胶增强填充用水合硅酸,其在橡胶中的分散性得到改善,耐磨耗性进一步提高,并且处理性能提高。
本发明所要解决的课题在于提供一种橡胶增强填充用水合硅酸,其在混配于橡胶中时的处理性能提高,在橡胶中的分散性得到改善,并且在混配有其的橡胶中作为橡胶增强性具有所期望的撕裂强度和耐磨耗性。
如上所述具有比较高的bet比表面积(例如230m2/g以上)的水合硅酸以往也获得了橡胶增强性,但本发明中,目的在于提供一种水合硅酸,该水合硅酸除了具有由bet比表面积得到的橡胶增强性以外,还具有通过水合硅酸在橡胶中的分散性得到改善而附加了功能的橡胶增强性、特别是耐磨耗性。
用于解决课题的手段
本发明人为了开发出下述水合硅酸进行了各种研究,该水合硅酸具有高bet比表面积(例如230m2/g以上),在混配到橡胶中时的处理性能提高,并且对于橡胶的分散性也提高。
本发明人特别分析了在将水合硅酸混配到橡胶中进行混炼时,随着混炼时间的经过,水合硅酸如何分散在橡胶中。基于该分析结果,进一步研究了水合硅酸具有何种颗粒结构和孔结构时能够解决上述课题。
其结果,发现下述水合硅酸作为同时满足高bet比表面积、高分散性、处理性能这三种独立性能的橡胶增强填充用水合硅酸是具有理想性能的水合硅酸,由此完成了本发明。该水合硅酸的bet比表面积为230~350m2/g,并且,
a):利用压汞法测定的孔半径1.9~100nm的孔容积(vhp-hg)为1.40~2.00cm3/g的范围;
b):利用氮气吸附脱附法得到的孔半径1.6~100nm范围的总孔容积(vn2)为1.60~2.20cm3/g的范围;
c):a)和b)的孔容积比vhp-hg/vn2为0.70~0.95的范围。
本发明如下所述。
[1]
一种橡胶增强填充用水合硅酸,其特征在于,bet比表面积为230~350m2/g的范围,并且,
a):利用压汞法测定的孔半径1.9~100nm的孔容积(vhp-hg)为1.40~2.00cm3/g的范围;
b):利用氮气吸附脱附法得到的孔半径1.6~100nm范围的总孔容积(vn2)为1.60~2.20cm3/g的范围;
c):a)和b)的孔容积比vhp-hg/vn2为0.70~0.95的范围。
[2]
如[1]所述的水合硅酸,其中,利用压汞法测定的孔半径100~1,000nm范围的孔容积为0.50~1.00cm3/g的范围。
[3]
如[1]~[2]中任一项所述的水合硅酸,其中,利用压汞法测定的孔半径1.6~62μm范围的孔容积为0.18~0.80cm3/g的范围,
利用压汞法测定的10~400kpa范围的滞后孔容积差为0.07cm3/g以上。
[4]
如[1]~[3]中任一项所述的水合硅酸,其中,在利用网眼200μm的筛进行分级时的筛余物量为整体的70重量%以上,并且颗粒硬度为5~35cn的范围。
[5]
如[1]~[4]中任一项所述的水合硅酸,其中,catb比表面积为200~350m2/g的范围。
[6]
如[1]~[5]中任一项所述的水合硅酸,其中,4重量%浆料的ph为5~8的范围,该浆料的滤液的电导率小于1,000μs/cm,含水率小于9%。
[7]
如[1]~[6]中任一项所述的水合硅酸,其中,上述水合硅酸为成型体。
发明的效果
根据本发明,可以提供一种水合硅酸,其在混配到橡胶中时的处理性能提高,并且除了通过使bet比表面积为规定范围而得到的橡胶增强性以外,还具有通过水合硅酸在橡胶中的分散性得到改善而附加了功能的优异的橡胶增强性、特别是耐磨耗性。
附图说明
图1示出实施例1中得到的水合硅酸的利用氮气吸附脱附法和压汞法测定的孔半径100nm以下的累积孔容积的比较。
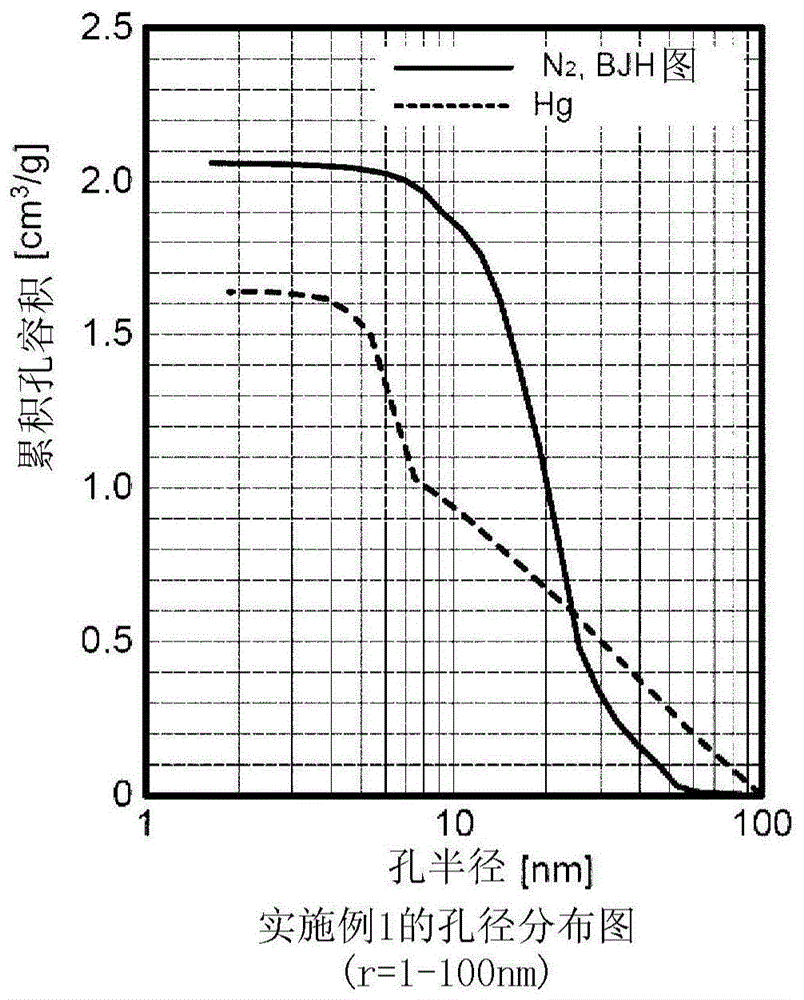
图2是包括实施例1中得到的压汞法1(测定范围r=1.9~6,400nm)的累积孔容积的孔分布图。
图3是包括实施例1中得到的氮气吸附脱附法(测定范围r=1.6~100nm)的累积孔容积的孔分布图。
图4是包括实施例1中得到的压汞法2(测定范围r=1.6~62μm[1,600~62,000nm])的累积孔容积的孔分布图。
具体实施方式
<水合硅酸>
本发明为一种橡胶增强填充用水合硅酸,其特征在于,
bet比表面积为230~350m2/g,并且,
a):利用压汞法测定的孔半径1.9~100nm的孔容积(vhp-hg)为1.40~2.00cm3/g的范围;
b):利用氮气吸附脱附法得到的孔半径1.6~100nm范围的总孔容积(vn2)为1.60~2.20cm3/g的范围;
c):a)和b)的孔容积比vhp-hg/vn2为0.70~0.95的范围。
本发明的水合硅酸中,bet比表面积小于230m2/g时,增强性如以往那样不充分。bet比表面积升高时,水合硅酸的制造趋于困难,实际上难以制造bet比表面积超过350m2/g的水合硅酸。bet比表面积的范围优选为230m2/g以上、更优选为240m2/g以上、进一步优选为250m2/g以上,优选为340m2/g以下、更优选为330m2/g以下、进一步优选为325m2/g以下。
a):利用压汞法,从100kpa使最高压力升压至400mpa,由此能够测定孔半径1.9nm~6,400nm范围的孔容积。本发明的水合硅酸利用压汞法测定的1.9nm以上100nm以下范围的孔容积(vhp-hg)为1.40~2.00cm3/g。利用压汞法进行的孔分布测定是对难以润湿试样的汞进行加压并由压入试样中的量求出孔分布的方法。只要没有特别声明,本发明的利用压汞法得到的孔分布是在使压力升压时得到的(从孔径大的一侧测定的)孔分布。
vhp-hg小于1.40cm3/g时,橡胶增强性不足;超过2.00cm3/g时,水合硅酸的实际制造趋于困难。vhp-hg优选为1.50cm3/g以上、更优选为1.60cm3/g以上1.95cm3/g以下、更优选为1.90cm3/g以下。
b):利用氮气吸附脱附法得到的孔半径1.6nm~100nm范围的总孔容积(vn2)为1.60~2.20cm3/g的范围。利用氮气吸附脱附法得到的孔分布测定是在液氮下(-196℃)、在真空状态至常压的范围使氮气分子吸附到试样表面,在常压至真空的范围使其脱附,由吸附脱附等温线测定孔分布的方法,可测定的孔半径范围为1.6nm~100nm。本发明的利用氮气吸附脱附法得到的孔分布是在脱附分布中得到的(从孔径大的一侧测定的)孔分布。
vn2小于1.60cm3/g时,橡胶增强性不足;vn2超过2.20cm3/g的情况下,水合硅酸的实际制造趋于困难。vn2优选为1.70cm3/g以上、更优选为1.80cm3/g以上,优选为2.15cm3/g以下、更优选为2.10cm3/g以下。
以往,vhp-hg≥vn2(vhp-hg/vn2≥1)的水合硅酸在通常的橡胶增强填充用水合硅酸、特别是bet比表面积230m2/g以上的橡胶增强填充用水合硅酸中是已知的。但是,vhp-hg≥vn2(vhp-hg/vn2≥1)的水合硅酸是在混炼到橡胶中时半径100nm以下的孔不会在测定压力下被压碎的程度的牢固的聚集体,因此即使分散于橡胶中,也作为大的聚集体而残留。其结果,橡胶分子不会侵入到聚集体的中心部,导致分散不良,难以得到充分的橡胶增强性。
在氮气吸附脱附法和压汞法的孔分布中,在两者共有的孔半径100nm以下的范围(微孔~大孔的范围)内进行比较的情况下,当水合硅酸的bet比表面积为230m2/g以上时,通常如上所述水合硅酸的内聚力增强。因此,即使在利用特别是最高以400mpa的压力压入的压汞法进行的测定中,半径100nm以下的孔也不会在压力下被压碎,与在真空至常压下测定的氮气孔容积相比,孔容积差也是同等的,或者检测到汞孔容积更大(vhp-hg≥vn2)。
与此相对,本发明的水合硅酸由于控制了聚集结构,因此在孔半径100nm以下的孔分布中,vhp-hg为1.40~2.00cm3/g的范围,vn2为1.60~2.20cm3/g的范围。但是,该孔容积范围内的聚集结构与现有的bet比表面积为230m2/g以上的水合硅酸相比较弱,因此聚集结构容易因利用压汞法测定时的压力而大幅压碎。因此,进一步具有下述特征c):a)和b)的孔容积比vhp-hg/vn2为0.70~0.95的范围。vhp-hg/vn2小于0.70的水合硅酸的制造困难,vhp-hg/vn2超过0.95的水合硅酸的内聚力强,在橡胶中的分散困难。
孔容积比vhp-hg/vn2优选为0.75~0.95、更优选为0.78~0.93的范围。
关于具有上述孔容积比vhp-hg/vn2的水合硅酸,若换用大孔和中孔(催化剂学会)的术语来说,推测大孔~中孔范围内的聚集体与现有的水合硅酸相比容易崩塌,因此,推测容易分散于橡胶中,具有优异的增强性。
本发明的水合硅酸优选利用压汞法测定的孔半径100~1,000nm范围(中压区域)的孔容积vmp-hg为0.50~1.00cm3/g的范围([2]所述的方式)。
孔容积vmp-hg小于0.50cm3/g时,具有发生下述任一种情况的倾向:该范围(大孔)内的聚集结构过强(以聚集不发生崩塌的水平被压紧的状态);或者由于聚集结构极弱,孔在汞的压力下被压碎而成块。因此,即使进行混炼而分散于橡胶中,在橡胶中作为水合硅酸聚集物存在的倾向也强,有时增强性的提高不充分。
相反地,对于孔容积vmp-hg超过1.00cm3/g的水合硅酸而言,水合硅酸多以微粒的状态存在。或者,堆积比重降低(变轻),在混炼中可能产生粉尘而导致作业环境恶化,有时向橡胶中的侵入也变得困难。因此,混炼时的初始的分散变差,未进行充分的分散,结果可能会导致增强性的降低。
本发明的水合硅酸优选利用压汞法测定的孔半径1.6~62μm范围(低压区域)的孔容积vlp-hg为0.18~0.80cm3/g的范围。此外,本发明的水合硅酸优选在利用压汞法升压至400kpa后、降压至10kpa时的孔容积(10~400kpa范围(低压区域)的滞后孔容积差)为0.07cm3/g以上([3]所述的方式)。
孔容积vlp-hg若为0.18cm3/g以上,压紧度不会过高,不会引起分散不良;孔容积vlp-hg若为0.80cm3/g以下,作为成型体是充分的,处理性能也良好。孔容积vlp-hg更优选为0.18~0.50cm3/g的范围。
滞后孔容积差若为0.07cm3/g以上,则聚集结构过强,在混炼到橡胶中时的初始阶段不会显示出不分散的状态(初期分散性良好)。滞后孔容积差更优选为0.10cm3/g以上。
本发明的水合硅酸优选在利用网眼200μm的筛进行分级时的筛余物量为整体的70重量%以上,并且颗粒硬度为5~35cn([4]所述的方式)。利用网眼200μm的筛进行分级时的筛余物量若为整体的70重量%以上,则处理性能良好,并且能够抑制粉尘的产生。颗粒硬度若为5cn以上,则能够防止粉尘产生,处理性能也良好,初始的分散性也良好。颗粒硬度若为35cn以下,则最终的分散性能也良好。
本发明的水合硅酸优选ctab比表面积为200~350m2/g的范围([5]所述的方式)。若为至少满足[1]所述的本发明条件的水合硅酸,在混配到橡胶中时可获得高增强性,除此以外,通过使ctab比表面积为上述范围,可确实地获得高增强性。需要说明的是,ctab比表面积超过350m2/g的情况下,有时水合硅酸本身的制造困难。ctab比表面积优选为220m2/g以上、更优选为240m2/g以上。
此外,本发明的水合硅酸中,bet比表面积与ctab比表面积这两个比表面积之比即bet/ctab比优选为1.0~1.2的范围。bet/ctab比若为1.2以下,则不会形成致密的聚集体,在橡胶中的分散性良好。若是bet/ctab比为1.0以上的水合硅酸,则可进行制造。
本发明的水合硅酸优选4重量%浆料的滤液的电导率小于1,000μs/cm,ph为5~8的范围([6]所述的方式),含水率小于9%。电导率若小于1,000μs/cm,则不易发生经时聚集,更优选小于800μs/cm、进一步优选小于500μs/cm。
4重量%浆料的ph范围5~8与通常的水合硅酸的ph相同。若ph为该范围内,则能够获得良好的硫化特性和增强性。
本发明的水合硅酸的含水率小于9%是适当的,含水率小于9%与通常的水合硅酸的含水率大致相同。含水率若为该范围内,则能够得到良好的硫化特性和增强性。电导率和ph可以通过制造工序中的水洗工序来控制,含水率可以通过干燥工序来控制。
<制造方法>
如上所述,本发明的水合硅酸是具有与迄今为止的水合硅酸不同的聚集结构的新型水合硅酸。通过例示对该水合硅酸的制法进行说明(本发明的方法称为多步反应)。
可以例示出基于下述多步反应的合成方法,可以通过包括i)~vii)的工序的合成方法来获得本发明的水合硅酸。其中,若在工序vi)中能够以某种程度进行水合硅酸的粒状化,则也可以省略压紧成型工序vii)。
i)将碱金属硅酸盐水溶液调整为规定量的ph(ph9.5~12)并预先投入反应槽中的工序;
ii)中和反应工序,包括同时滴加碱金属硅酸盐水溶液和无机酸的工序和不添加碱金属硅酸盐水溶液而仅滴加无机酸的工序;
iii)中和反应工序,其为同时滴加碱金属硅酸盐水溶液和无机酸的工序,其中,各自的流量独立地为工序ii)的同时滴加工序中的流量的20~80%的范围;
iv)不添加碱金属硅酸盐水溶液而仅滴加无机酸,使ph<7后停止中和反应的工序。
其中,工序i)~iv)是一边对反应浆料进行搅拌或/和循环,一边维持70~90℃的范围来实施的,工序ii)~iii)是一边维持ph9.5~12的范围一边来实施的。
v)对所得到的水合硅酸浆料进行过滤,并且利用与所得到的水合硅酸的滤饼相同量以上的水进行水洗的工序;
vi)调整水合硅酸的含水率的干燥工序;
vii)对所得到的水合硅酸进行压紧成型的工序。
碱金属硅酸盐水溶液没有特别限定,例如可以使用市售的硅酸钠。无机酸也没有特别限定,优选为硫酸,浓度可以使用稀硫酸~浓硫酸中的任一种。
工序i)~ii)是用于使bet比表面积为230m2/g以上的准备工序和形成半径为100nm以下的孔容积的准备工序。工序i)中例如利用水对市售的碱金属硅酸盐水溶液进行稀释并调整为规定的ph。只要能够将ph维持为规定范围,是否添加盐类等其他化合物均可。
工序ii)中的同时滴加碱金属硅酸盐水溶液和无机酸的工序、和不添加碱金属硅酸盐水溶液而仅滴加无机酸的顺序为不同顺序。例如,不添加碱金属硅酸盐水溶液而仅滴加无机酸的工序在工序ii)的前半部分、中间、后半部分中的任一者实施即可。
工序iii)是现有方法中没有的本发明中新增加的工序。通过增加该工序,能够在形成半径为100nm以下的孔的结构性的同时,控制半径超过100nm且为1,000nm以下的孔容积及其结构性。工序iii)例如可以以相对于工序ii)的同时滴加的工序为20~80%的范围中的任一恒定流量来实施,也可以在工序的过程中在上述范围内至少改变1次流量来实施。从获得具有所期望的结构性的水合硅酸的方面考虑,碱金属硅酸盐水溶液和无机酸的流量优选为工序ii)中的碱金属硅酸盐水溶液和无机酸的流量的20~60%的范围。
推荐工序ii)和工序iii)合计以80~140分钟左右、优选100~130分钟左右完成的方法。需要说明的是,工序ii)和工序iii)也可以重复进行2次以上。即,例如可以实施第1次的工序ii)和工序iii),接下来,不进入工序iv)而再次实施工序ii)和工序iii),之后进入工序iv)。工序ii)和工序iii)的重复例如可以进行2次、3次或4次。通过重复进行工序ii)和工序iii),具有容易获得具有所期望的结构性的水合硅酸的优点。
本发明的水合硅酸在中和反应工序中直至停止反应为止(直至上述工序iii)为止)始终保持ph为7以上、优选保持ph为10.0~12.0、更优选保持ph为11.0~12.0。此外,中和反应中,进行浆料的搅拌、循环中的一者或两者是适当的。特别是在搅拌速度、循环速度快的情况下,能够抑制水合硅酸的聚集,对于本发明的水合硅酸的制造有利。
工序iv)的停止中和反应的工序通过停止碱金属硅酸盐水溶液的添加并继续仅滴加无机酸来实施。继续仅滴加无机酸进行至反应液(浆料)达到ph<7是适当的。
工序v)的水合硅酸的过滤、水洗例如可以利用压滤机等来进行,也可以使用使其带有压榨功能的压滤机,在进行水洗后实施压榨。
工序vi)是将含水率控制为规定数值的干燥工序。干燥工序的方法没有特别限定,具体而言,可以为静置干燥方式,除了上述喷雾干燥机以外,还可例示出利用旋转闪蒸干燥机等市售的干燥机进行控制的方法。在通过这些之中的任一种干燥方法实施干燥并且还进行了粒状化的情况下,可以省略工序vii)的压紧成型。粒状化可以根据干燥方法适当设定条件来实施。
(添加剂的使用)
水洗后,可以添加用于提高橡胶增强性的添加剂(预先添加铝、表面活性剂、硅烷偶联剂等)。但是,本发明的水合硅酸即使完全不添加这些添加剂也容易获得高增强性,因此添加剂优选在后述的橡胶混配时添加。在不得不添加添加剂的情况下,例如可以例示下述方法:向工序v)中得到的水合硅酸滤饼中加入水,再次制成浆料后,加入添加剂并利用喷雾干燥机等进行干燥。
工序vii)是对干燥工序中得到的水合硅酸进行压紧成型的工序。通过进行压紧成型,能够控制半径1.6μm~62μm(1,600~62,000nm)的孔容积及其结构性。如上所述,尽管可以仅通过上述工序vi)来进行控制,但从能够稳定地控制结构性的方面考虑,优选对干燥后的水合硅酸进行压紧成型。此外,通过进行压紧成型,能够使利用网眼200μm以上的筛进行分级时的筛余物量为整体的70重量%以上、颗粒硬度为5~35cn,从该方面考虑也是优选的。
具体的压紧成型方法可以使用市售的利用干式法的成型机,但没有特别限制。压紧成型方法大致可分成混合成型法、强制成型法和热利用成型法这3种,本发明中,特别优选使用强制成型法。
强制成型法进一步包括压缩辊、压块辊、压片等压紧成型法或使用螺杆等的挤出成型法等,本发明中,特别优选使用压紧成型法。另一方面,压片机等过度的压紧成型机会过度地破坏水合硅酸的孔结构,因此也有可能无法达到本发明的目的,不优选。
在市售的辊式成型机的情况下,水合硅酸的粉体一边通过进料螺杆的旋转被加压一边被压入到旋转的压辊间而被压缩,压紧成型为板状或粒状。压辊包括平滑、带槽、带波纹的压辊等,本发明中可以使用任一种辊。无论何种情况下,均对辊形状、速度、压力等进行调整以达到上述说明的适度的孔和粒度、颗粒硬度来使用。
粒状化、压紧成型等方法是为了防止水合硅酸的粉尘产生或进一步提高处理性能而通常对橡胶增强填充用的水合硅酸进行的方法。
本发明的水合硅酸尽管bet比表面积为230m2/g以上,但即使在一定条件下进行粒状化或压紧成型等加工,半径1,000nm以下(微孔~大孔)的孔容积也很少变化,因此能够维持良好的橡胶增强性。因此,即使在适度的一定条件下进行压紧成型或粒状化,也仅在防止粉尘产生或提高处理性能的方面发挥作用,橡胶增强性不会降低。
<橡胶混配>
本发明的水合硅酸能够用作各种橡胶组合物的增强填充用途,橡胶组合物的用途不仅包括轮胎,还包括轮带等工业用部件。可使用(混配)本发明的水合硅酸的橡胶组合物没有特别限制,作为橡胶,可以是单独包含天然橡胶(nr)或二烯系合成橡胶或者共混地包含它们的橡胶组合物。作为合成橡胶,可以举出例如合成聚异戊二烯橡胶(ir)、聚丁二烯橡胶(br)、丁苯橡胶(sbr)、丁腈橡胶(nbr)、丁基橡胶(iir)等。
相对于天然橡胶和/或二烯系合成橡胶100重量份,本发明的水合硅酸例如可以混配5~100重量份。但是,并没有限定于该范围的意图。
<橡胶混配时的添加剂>
上述橡胶组合物也可以在橡胶混配时根据需要(除了添加到水合硅酸中以外)加入铝化合物、硅烷偶联剂、表面活性剂等在橡胶组合物中使用的各种添加剂。
在将本发明的水合硅酸用于橡胶组合物的情况下,除了上述的橡胶(添加剂)以外,可以根据需要适当混配炭黑、软化剂(蜡、油)、抗老化剂、硫化剂、硫化促进剂、硫化促进助剂等通常在橡胶工业所用的混配剂。橡胶组合物可以利用混合辊或班伯里混炼机等市售的混炼机将上述橡胶成分、水合硅酸、硅烷偶联剂、上述根据需要混配的上述炭黑、橡胶混配剂等混配来制备。
混炼后的橡胶组合物在成型为所期望的形状后,进行硫化而成为各种橡胶产品。
混配有本发明的水合硅酸的橡胶组合物可以适合用于轮胎、传送带、软管等橡胶制品中,作为产品的轮胎、传送带、软管等橡胶制品的增强性、高耐磨耗性等优异。另外,使用了混配有本发明的水合硅酸的橡胶组合物的充气轮胎能够将上述橡胶组合物用于轮胎胎面部,可得到轮胎胎面部的增强性、高耐磨耗性优异的充气轮胎。
实施例
下面,基于实施例进一步详细地说明本发明。但是,实施例为本发明的例示,并没有将本发明限定于实施例的意图。
■水合硅酸的各物性的测定法
●bet比表面积(n2法比表面积)
使用全自动比表面积测定装置(型号:macsorb(r)hm1201型;mountech公司制造),通过1点法进行测定。
●ctab比表面积
基于jisk6430(橡胶用混配剂-二氧化硅-试验方法)进行测定。其中,将ctab分子的吸附截面积设为
●利用氮气吸附脱附法得到的孔分布
使用高精度气体/蒸气吸附量测定装置(型号:belsorpmax;日本bel公司制造),通过barret-joyner-halenda法(bjh法)测定了孔半径1.6~100nm范围的总孔容积(vn2)。需要说明的是,测定结果为脱附侧的(从孔容积大的一侧测定的)孔分布和孔容积。
●利用压汞法得到的孔分布1[孔半径1.9~1,000nm]
使用压汞仪(型号:pascal440;thermoquest公司制造)从100kpa升压至最高压力400mpa,测定了孔半径1.9~6,400nm范围的汞孔分布和孔容积。需要说明的是,测定结果是在升压时(从孔容积大的一侧)测定的孔分布和孔容积。利用附带的软件分析所得到的测定结果,进行1.9~100nm(vhp-hg)、100~1,000nm的孔容积以及与利用氮气吸附脱附法得到的孔容积(vn2)的孔容积比vhp-hg/vn2的比例计算等。
●利用压汞法得到的孔分布2[孔半径1.6~62μm(1,600~62,000nm)]
使用压汞仪(型号:pascal140;thermoquest公司制造)从10kpa升压至400kpa,测定了孔半径1,600~62,000nm(或1.6~62μm)范围的汞孔。进而还测定了升压至400kpa后降压至10kpa时的孔容积(滞后孔容积差)。对成型后的1.5mm左右的水合硅酸进行了测定。
●ph的测定
基于jisk5101-17-2(颜料试验方法),对于用纯水调整为4重量%的浆料的ph,利用市售的玻璃电极ph计(型号:f-53;堀场制作所公司制造)测定了指示值稳定时的值。
●电导率
将4g的水合硅酸(在105℃干燥2小时后的加热失重为6%以下)添加到50ml的蒸馏水中,充分混合后,进行5分钟煮沸处理。之后,使用蒸馏水将总容量调整为100ml后,进行过滤,利用电导率计(型号:cm30r东亚dkk公司制造)对该滤液进行了测定。
●含水率
基于jisk5101-15-1(颜料试验方法),由105℃干燥2小时后的失重值求出。
●颗粒硬度
基于jisk6219-3(橡胶用炭黑-造粒颗粒的特性)的颗粒硬度b法,按照网眼1.4mm、1.7mm的筛的顺序重叠,将试样载置于1.7mm的筛上,置于振荡机中。之后,对1.4mm的筛上的20个造粒颗粒进行测定,求出平均值作为造粒颗粒的硬度。
●筛余物
基于jisk0069(化学产品的筛分试验方法)的干式筛分试验方法的手动筛分,使用网眼200μm的筛并使筛倾斜20度,以1分钟120次的比例用手敲打筛,由筛上的筛余物重量算出。
■橡胶混配物制备法和相关的各物性的测定法
●混配物制备法1
按照表1所示的配方,通过下述混炼过程制备出橡胶试验用样品。将市售的nbr(丁腈橡胶)100重量份卷绕到8英寸开炼机上,进行3分钟塑炼后,用时35分钟依次投入混配物a。将该母料在室温下冷却后,再次用8英寸开炼机充分混炼混配物b的水合硅酸30重量份,得到未硫化的橡胶组合物。
●粉尘浓度测定
将水合硅酸混炼到母料中时,在8英寸开炼机的正上方30cm处设置数字粉尘计(型号:ld-5d;柴田科学公司制造),在将水合硅酸30重量份投入到母料中时进行粉尘测定。由利用数字粉尘计测定的粉尘相对浓度(cpm)计算出质量浓度换算系数(k值、粉尘浓度=k值×cpm),并换算成粉尘浓度(g/m3)。
【表1】
※表中的单位为phr(相对于聚合物100重量份的重量份)
●混配物制备法2
按照表2所示的配方,通过下述混炼过程制备出橡胶试验用样品。
(i)利用1.7l班伯里混炼机(神户制钢公司制造)对聚合物700g进行30秒塑炼,加入表2的混配物a,通过上顶栓压力或转速按照取出时的混合物温度为140~150℃的方式进行调节,混炼5分钟后取出。
(ii)将混合物在室温下冷却后,再次利用1.7l班伯里混炼机加入表2的混配物b,混炼50秒后取出(取出时的温度设定为100℃以下),利用8英寸开炼机进行压片,得到未硫化的橡胶组合物。
【表2】
※表中的单位为phr(相对于聚合物100重量份的重量份)
※硅烷偶联剂的混配量(变量)=ctab比表面积×0.029-0.185
●关于混配物制备法1和2
混配物制备法1和2不能一概地进行比较,但就混配时的机械强度(混配物的份额)而言是混配物制备法1较弱,容易判断出差异。另外,混配物制备法1与工业用橡胶等中使用的配方比较接近,混配物制备法2与轮胎的胎面部分使用的配方接近。由于橡胶物性根据配方而不同,因此本发明中进行了各自的混配试验,也确认到了因混配种类的差异所致的物性变化。
●硫化
将未硫化的橡胶组合物放入试片成型模具中,利用蒸气硫化压力机(末次铁工所公司制造)在150℃的温度、4.0~18mpa的压力下进行10~20分钟硫化,得到试片。
硫化中使用的试片成型模具如下。
●撕裂强度测定用模具(型号:mp-124njkac;dumbbell公司制造)
●分散性测定用模具(型号:mpa-609ak;dumbbell公司制造)
●磨耗试验用模具(型号:mpl-309lakc;dumbbell公司制造)
●分散性评价
基于iso11345,使用dispergrader1000(optigrade公司制造)对硫化后的试片截面进行测定。分散度以x值进行评价。值越大则分散性越好,以a、b、c、d四个等级进行评价。在混配物制备法1中,将x值为8.5以上记为a,8.0~8.5记为b,7.5~8.0记为c,小于7.5记为d。另外,在混配物制备法2中,将x值为8.0以上记为a,7.5~8.0记为b,7.0~7.5记为c,小于7.0记为d。无论哪种情况,均将b以上作为合格。
●撕裂强度
基于jisk6252-1(硫化橡胶和热塑性橡胶-撕裂强度的求法-)的试验方法b,利用剪裁机将硫化橡胶片冲切成角型(无缺口),利用schopper型拉伸试验机(岛津制作所公司制造)拉伸至试片断裂为止,求出撕裂强度。测定结果以将参考例1作为100(基准)时的指数来求出。指数越高,表示撕裂强度越高,以a、b、c、d四个等级进行评价。在混配物制备法1中,将撕裂强度比基准提高15%以上时(指数115以上)记为a,提高5~10%时(指数105~110)记为b,与基准同等时(指数95~105)记为c,与基准相比变差时(指数小于95)记为d。另外,在混配物制备法2中,将撕裂强度比基准提高10%以上时(指数110以上)记为a,提高5~10%时(指数105~110)记为b,与基准同等时(指数95~105)记为c,与基准相比变差时(指数小于95)记为d。无论哪种情况,均将b以上作为合格。
●磨耗试验
基于jisk6264-2(硫化橡胶和热塑性橡胶-耐磨耗性的求法-),利用akron型磨耗试验机进行测定。设置直径
[实施例1]
经由以下的i)至vii)的工序制造水合硅酸,并进行了评价。需要说明的是,下述工序i)~iv)是在具备搅拌机和循环泵的240升不锈钢容器中,在温度保持为76℃的情况下一边始终进行搅拌和循环一边实施的。另外,作为以下记载的硅酸钠水溶液,使用sio2浓度12.8wt%、na2o浓度4.0wt%、sio2/na2o摩尔比3.2的3号硅酸钠,作为硫酸,使用95wt%的浓硫酸。
i)在温水80升中加入硅酸钠水溶液,直至ph达到11.5为止。
ii-1)同时滴加硫酸1.06升和硅酸钠水溶液35分钟,调整硅酸钠水溶液的流量以维持ph为11.5。
ii-2)接着,停止硅酸钠水溶液的滴加,仅滴加硫酸直至ph达到11.0为止。
ii-3)再次在维持ph为11.0的情况下以与工序ii-1)相同条件的流量同时滴加硅酸钠水溶液和硫酸15分钟。
iii)调整流量以使硅酸钠水溶液和硫酸的流量相对于工序ii-1)和ii-3)为43%,在维持ph为11.0的情况下用60分钟同时滴加。
iv)停止硅酸钠水溶液的滴加,仅滴加硫酸来降低ph,在ph达到3.0后也停止硫酸的滴加,完全终止中和反应,得到水合硅酸浆料。
v)用压滤机对所得到的水合硅酸浆料进行过滤,还进行水洗,得到水合硅酸滤饼。
vi)对于水合硅酸滤饼,按照sio2浓度为120g/升的方式再次加入水,进行浆料化后,利用喷雾干燥机(型号:an-40r型ashizawaniroatomizer公司制造)按照含水率小于9%的方式进行干燥。
vii)对于所得到的干燥粉,利用辊压式成型机(辊式压实机、型号:fr125×40型turbokogyo公司制造)和附带的螺杆加料器,以加料量11.3kg/h、辊间隔1.05mm、压缩压力0.3吨/cm、辊转速8rpm进行混合成型,得到水合硅酸成型体。
[实施例2]
经由下述i)至vii)的工序制造水合硅酸,并进行了评价。需要说明的是,装置、硅酸钠水溶液、硫酸和温度、以及至工序i)为止的操作在与实施例1相同的条件下进行。
i)在温水80升中加入硅酸钠水溶液,直至ph达到11.5为止。
ii-1)接着,仅滴加硫酸,使ph降低至11.0为止。
ii-2)同时滴加硫酸0.76升和硅酸钠水溶液25分钟,调整硅酸钠水溶液的流量以维持ph为11.0。
iii-1)调整流量以使硅酸钠水溶液和硫酸的流量相对于工序ii-2)为43%,在维持ph为11.0的情况下用25分钟同时滴加。
ii-3)再次在维持ph为11.0的情况下以与工序ii-2)相同条件的流量同时滴加硅酸钠水溶液和硫酸20分钟。
iii-2)接着,在维持ph为11.0的情况下以与工序iii-1)相同条件的流量同时滴加硅酸钠水溶液和硫酸40分钟。
之后,在与实施例1相同的条件下进行工序iv)~vi)。
vii)将vi)中得到的干燥粉的加料量增加至11.5kg/h,除此以外在与实施例1相同的条件下成型,得到水合硅酸成型体。
[实施例3]
经由下述i)至vii)的工序制造水合硅酸,并进行了评价。需要说明的是,除了使温度为81℃以外,装置、硅酸钠水溶液、硫酸、以及至工序i)为止的操作在与实施例1相同的条件下进行。
i)在温水80升中加入硅酸钠水溶液,直至ph达到11.5为止。
ii-1)同时滴加硫酸0.61升和硅酸钠水溶液20分钟,调整硅酸钠水溶液的流量以维持ph为11.5。
ii-2)接着,停止硅酸钠水溶液的滴加,仅滴加硫酸直至ph达到11.0为止。
ii-3)再次在维持ph为11.0的情况下以与工序ii-1)相同条件的流量同时滴加硅酸钠水溶液和硫酸30分钟。
iii)调整流量以使硅酸钠水溶液和硫酸的流量相对于工序ii-1)和ii-3)为43%,在维持ph为11.0的情况下用60分钟同时滴加。
之后,在与实施例1相同的条件下进行工序iv)~vi)。
vii)将vi)中得到的干燥粉的加料量增加至13.2kg/h,除此以外在与实施例1相同的条件下成型,得到水合硅酸成型体。
[实施例4]
经由下述i)至vii)的工序制造水合硅酸,并进行了评价。需要说明的是,除了使温度为85℃以外,装置、硅酸钠水溶液以及硫酸在与实施例1相同的条件下进行。
i)在温水80升中加入硅酸钠水溶液,直至ph达到11.7为止。
ii-1)接着,仅滴加硫酸,使ph降低至11.0为止。
ii-2)同时滴加硫酸1.52升和硅酸钠水溶液50分钟,调整硅酸钠水溶液的流量以维持ph为11.0。
iii)调整流量以使硅酸钠水溶液和硫酸的流量相对于工序ii-2)为43%,在维持ph为11.0的情况下用60分钟同时滴加。
之后,在与实施例1相同的条件下进行工序iv)~vi)。
vii)将vi)中得到的干燥粉的加料量增加至11.6kg/h,除此以外在与实施例1相同的条件下成型,得到水合硅酸成型体。
[实施例5]
经由下述i)至vii)的工序制造水合硅酸,并进行了评价。需要说明的是,除了使温度为87℃以外,装置、硅酸钠水溶液以及硫酸在与实施例1相同的条件下进行。
i)在温水80升中加入硅酸钠水溶液,直至ph达到11.6为止。
ii-1)同时滴加硫酸1.06升和硅酸钠水溶液35分钟,调整硅酸钠水溶液的流量以维持ph为11.5。
ii-2)接着,停止硅酸钠水溶液的滴加,仅滴加硫酸直至ph达到11.0为止。
ii-3)再次在维持ph为11.0的情况下以与工序ii-1)相同条件的流量同时滴加硅酸钠水溶液和硫酸15分钟。
iii)调整流量以使硅酸钠水溶液和硫酸的流量相对于工序ii-1)和ii-3)为43%,在维持ph为11.0的情况下用60分钟同时滴加。
之后,在与实施例1相同的条件下进行工序iv)~vi)。
vii)将vi)中得到的干燥粉的加料量增加至11.7kg/h,除此以外在与实施例1相同的条件下成型,得到水合硅酸成型体。
[参考例1]
作为具有220m2/g以上的bet比表面积且作为橡胶增强填充剂用水合硅酸在市售的水合硅酸的例子,使用nipsilkq(tosohsilica公司制造)进行了评价。需要说明的是,橡胶混配物的耐磨耗性、撕裂强度等各物性评价以该nipsilkq的物性值为基准(100),以指数进行了比较。
[比较例1]
按照与日本特表2017-514773号公报(专利文献1)的实施例5中记载的方法相同的配方制造二氧化硅滤饼,得到具有250m2/g的bet比表面积的实质上为球形的珠粒形态的水合硅酸。
需要说明的是,制造中使用与实施例1相同的装置(反应容器),以相对于日本特表2017-514773的实施例5为11分之1的处理量进行水合硅酸的中和合成,用压滤机进行过滤,在得到二氧化硅滤饼后,添加规定量的铝酸钠溶液和中和溶液,除此以外利用实施例1的工序vi)的方法按照含水率小于9%的方式进行干燥。但是,根据现有文献1的方法,未进行实施例1的工序vii)的操作。
[比较例2]
使用与实施例1相同的反应容器,按照与日本特开平10-194722号公报的实施例1中记载的方法相同的配方,通过箱式干燥的方法获得了水合硅酸。需要说明的是,制造以相对于日本特开10-194722的实施例1为1.2倍的处理量来实施。
[比较例3]
使用与实施例1相同的反应容器,按照与日本特开2012-17440号公报的制造例2中记载的方法相同的配方,制作出水合硅酸,之后在与实施例4的工序vii)相同的条件下进行水合硅酸的成型,得到水合硅酸成型体。需要说明的是,制造以相对于日本特开2012-17440的制造例2为1.3倍的处理量来实施。
比较例1~3的制造方法中不存在与实施例1~5的工序iii)相当的工序。此外,比较例1和2的制造方法中,也不存在与实施例1~5的工序vii)相当的工序。
■结果说明<孔>
图1中示出实施例1中得到的水合硅酸的利用氮气吸附脱附法和压汞法测定的孔半径小于100nm的累积孔容积的比较。
由比较结果可知,实施例1的水合硅酸的小于100nm的孔在利用压汞法测定时(vhp-hg)的孔容积小于利用氮气吸附脱附法测定(vn2)的孔容积(vhp-hg<vn2)。
另外,图2~4中示出包括实施例1中得到的氮气吸附脱附法(测定范围r=1.6~100nm)、压汞法1(测定范围r=1.9~6,400nm)、压汞法2(测定范围r=1.6~62μm[1,600~62,000nm])的累积孔容积的孔分布图。
由这些图和测定数据求出利用氮气吸附脱附法得到的孔容积(vn2)、利用压汞法得到的孔半径1.9~100nm范围的孔容积(vhp-hg)和100~1,000nm范围的孔容积、利用压汞法得到的孔半径1.6~62μm[1,600~62,000nm]范围的孔容积和滞后孔容积差、即在进行升压后进行降压时的孔容积。
■结果说明(表)
将实施例1~5、比较例1~3的水合硅酸的物性示于表3,将橡胶混配试验结果示于表4。
【表3-1】
实施例的水合硅酸物性
【表3-2】
参考例、比较例的水合硅酸物性
【表4】
利用混配物制备法1、混配物制备法2的橡胶混配试验结果
■考察(说明)
表3-1的实施例的水合硅酸均为(vhp-hg<vn2),处于vhp-hg/vn2=0.70~0.95的范围。
在表4的耐磨耗性、撕裂强度的评价中,混配物制备法1、2与比较例相比均确认到显著的改善效果。
特别是耐磨耗性的改善效果显著,同时不仅增强性提高,起粉也少,还维持了处理性能。
参考例1由于是微粒的颗粒状,因此无法测定颗粒硬度,但粉尘浓度低,是作业性良好的市售的水合硅酸。(橡胶物性评价基准)
比较例1是bet比表面积为230m2/g以上、但vhp-hg/vn2=0.99的水合硅酸。若能进行压紧成型,则有可能能够改善粉尘浓度等,但预测其在橡胶中的分散性、增强性进一步降低,因此在未进行压紧成型的情况下评价了橡胶物性。
比较例2的vhp-hg/vn2高达0.99。此外,ctab比表面积也低,因此未得到高增强性(特别是耐磨耗性)。另外,若能进行压紧成型,则有可能能够改善粉尘浓度等,但由于vhp-hg/vn2=0.99,因此能够显著改善橡胶增强性(特别是耐磨耗性)的可能性低。
比较例3是通过进行成型而使混炼时的粉尘浓度少、处理性能优异的bet比表面积为230m2/g以上的水合硅酸,但vhp-hg/vn2=1.01,在橡胶中的分散不充分,增强性也不充分。
综上,实施例1~5的水合硅酸由于具有高比表面积且处于vhp-hg/vn2=0.70~0.95的范围,因此在混炼到橡胶中时可得到高耐磨耗性和高撕裂强度。另外可知,还兼具即使成型也容易分散的特征,是作业性也得到改善的优异的水合硅酸。特别是实施例1为vhp-hg/vn2=0.80这样的低值,耐磨耗性提高的效果显著。
如以上所说明,本发明的水合硅酸尽管bet比表面积为230~350m2/g,但孔半径100nm以下的孔与现有的水合硅酸相比容易崩塌,孔半径100~1,000nm、1.6μm以上的孔也具有适度的结构性,因此能够在提高处理性能的状态下混配到橡胶中,混配时可良好地分散,获得高增强性(特别是耐磨耗性)。
因此,本发明能够提供一种水合硅酸,其在混配到橡胶中时的处理性能提高,并且除了通过使bet比表面积为规定范围而得到的橡胶增强性以外,还具有通过水合硅酸在橡胶中的分散性得到改善而附加了功能的优异的橡胶增强性、特别是耐磨耗性。
工业实用性
本发明在涉及水合硅酸的技术领域中有用。
- 还没有人留言评论。精彩留言会获得点赞!