一种Y型多缩乙二醇衍生物及其制备方法与流程
本发明涉及一种Y型多缩乙二醇衍生物及其制备方法,特别是,本发明涉及一种以N为分支节点的具有支化结构的多缩乙二醇衍生物。
背景技术:
聚乙二醇(PEG)被认为是已知聚合物中蛋白和细胞吸收水平最低的聚合物,而且,其具有良好的水溶性和生物相容性,无毒,无免疫性,无致畸性且无抗原性的优势,被广泛应用于药物修饰、制剂制备与医疗器械材料等领域。从1991年开始,第一种用PEG修饰的药物PEG-ADA被FDA批准上市后,各大制药公司对PEG在药物领域的研发投入了极大的兴趣。近几年来,上市的产品有PEG-生长抑素、PEG-干扰素、PEG-粒细胞集落因子等。目前,尚有几十种PEG修饰药物处于研究或临床试验阶段。
虽然PEG在药物修饰等领域具有独特的优势,但其还存在一些不足之处:1)PEG是聚合产物,是一种混合物,其本身存在不均一的缺陷,这不但限制了PEG的应用,如在用PEG对超氧化物歧化酶进行修饰时,会产生一系列不均一的产物(Characterization of the heterogeneity of polyethylene glycol-modified superoxide dismutase by chromatographic and electrophoretic techniques,JSnider et.al.,Journal of Chromatography A 1992,599,141-155);而且给药物合成时不同批次产物的统一性以及最终的产品检测带来了一定困难;2)线性PEG分子本身分子量较高,但其只有两端可以连接药物,不可避免的造成了载药量的下降。
为了克服PEG所带来的不利影响,Dhawan等人(Synthesis of polyamide oligomers based on 14-amino-3,6,9,12-tetraoxatetradecanoic acid,Dhawan et.al.,Bioconjugate Chemistry 2000,11,14-21)用有机合成的方法,通过醇钠与磺酸酯的反应制备了与PEG结构相同的线性多缩乙二醇(即有确定分子量的PEG)及其衍生物。多缩乙二醇与PEG具有相同的化学式,但两者并不一致,例如PEG12与EG12(十一缩十二乙二醇),前者是EG11、EG12、EG13及更长或更短的多缩乙二醇所组成的混合物,而后者是一个确定的化合物,只含有EG12。
线性多缩乙二醇虽然解决了PEG本身不均一的缺陷,但其本身又带来了新的问题。其制备方式需要首先保护多缩乙二醇的一端,然后在严格的无水条件下进行Williamson合成,然后脱保护得到更长链段多缩乙二醇。该合成方法步骤较多,操作复杂,而且条件苛刻,需要用到严格的无水条件得到醇钠且脱保护时需用氢气还原,限制了其进一步推广 和大面积的使用,而且,链段较短的直链多缩乙二醇的负载量虽然有所提高,但水溶性也因此显著降低,使其应用范围大大缩减。
为解决现有技术中的缺陷,本发明提供了一种Y型多缩乙二醇衍生物及其制备方法。
技术实现要素:
本发明的一个目的是提供一种Y型多缩乙二醇化合物,能够解决现有聚乙二醇产物不均一的问题,同时可以增加药物的负载量。
本发明的另一个目的是解决了多缩乙二醇修饰不溶性药物时由于负载量的提高造成水溶性不足的问题。
本发明的还一个目的是提供一种多缩乙二醇化合物的制备方法,其步骤简单,条件温和,不需要严格的无水环境,也不需要进行保护和脱保护的步骤。
因而,本发明一方面提供了一种Y型多缩乙二醇衍生物,其具有式(Ⅰ)的结构:
其中:
m,n为0-30的整数;
A、B为相同或不同的Y-X-结构;
X为连接基团,选自:-(CH2)i-、-(CH2)iNH-、-(CH2)iOCOO-、-(CH2)iOCONH-、-(CH2)iNHCONH-、-OC(CH2)iCOO-、-(CH2)iCOO-、-(CH2)iCONH-,i为从0-10的整数;
Y为活性端基,选自:C1-C6的烷氧基、羟基、氨基、氨甲基、马来酰亚胺、羧基、巯基、琥珀酰亚胺碳酸酯、琥珀酰亚胺乙酸酯、琥珀酰亚胺丙酸酯、琥珀酰亚胺琥珀酸酯、琥珀酰亚胺、二硫吡啶基、丙酸基、醛基、正吡啶二硫基、巯酯基、丙烯酸基、叠氮基、戊二酸基、酰肼基、炔基、对硝基苯碳酸酯基、异氰酸基、邻二硫吡啶基、硅烷基、羧甲基;
E1为多缩乙二醇基团,其结构为:(CH2CH2O)j,j为0-100的整数;
E2为多缩乙二醇基团,其结构为:(OCH2CH2)k,k为0-100的整数。
优选地,本发明所述的式(Ⅰ)的结构中,m为2-10的整数,n为2-10的整数;更为优选的,m为2、3、4或5,n为2、3、4或5。
优选地,本发明所述的式(Ⅰ)的结构中,连接基团X中i为0-6的整数,更优选的i为0、1、2、3或4;连接基团X优选为-(CH2)i-、-(CH2)iNH-、(CH2)iCONH-。
优选地,本发明所述的式(Ⅰ)的结构中,活性基团Y选自:甲氧基、羟基、氨基、 巯基、羧基、酯基、醛基、丙烯酸基或马来酰亚胺基。
优选地,式(Ⅰ)的结构中,多缩乙二醇基团E1中j为0-20的整数,更优选地,j为1-12的整数,最为优选的j为1、2、3、4、5、6、7或8。
优选地,式(Ⅰ)的结构中,多缩乙二醇基团E2中k为0-20的整数,更优选地,k为0-12的整数,最为优选的k为0、1、2、3、4、5、6、7或8。
在本发明的一个实施方式中,所述的Y型多缩乙二醇衍生物具有以下的结构(Ⅱ):
在本发明的一个实施方式中,所述的Y型多缩乙二醇衍生物具有以下的结构(Ⅲ):
在本发明的一个实施方式中,所述的Y型多缩乙二醇衍生物,具有以下的结构(Ⅳ):
在本发明的一个实施方式中,所述的Y型多缩乙二醇衍生物,具有以下的结构(Ⅴ):
在本发明的一个实施方式中,所述的Y型多缩乙二醇衍生物,具有以下的结构(Ⅵ):
在本发明的一个实施方式中,所述Y型多缩乙二醇衍生物具有以下的结构(Ⅶ):
在本发明的一个实施方式中,所述的Y型多缩乙二醇衍生物具有以下的结构(Ⅷ):
本发明的另一方面提供了一种式(Ⅰ)结构的Y型多缩乙二醇衍生物的制备方法,其步骤包括:
(1)将端基已修饰的多缩乙二醇衍生物,例如是甲氧基多缩乙二醇,进行卤化或磺酸酯化;(2)将步骤(1)的产物与一端端基为氨基的多缩乙二醇衍生物反应,(3)任选的,对步骤(2)产物的端基基团进行改造制备得到结构。
本发明所述Y型多缩乙二醇衍生物的制备方法步骤(1)中,所述的端基已修饰的多缩乙二醇衍生物,其结构为Z-X-E1-(CH2)n-OH,
其中:
n为0-30的整数;
X为连接基团,选自:-(CH2)i-、-(CH2)iNH-、-(CH2)iOCOO-、-(CH2)iOCONH-、-(CH2)iNHCONH-、-OC(CH2)iCOO-、-(CH2)iCOO-、-(CH2)iCONH-,i为从0-10的整数;
Z为本发明所述的式(Ⅰ)结构中Y所定义的活性端基或者Z选自以下基团:甲酯、乙酯、叔丁酯、缩醛基、苄氧基;
所述Y为:C1-C6的烷氧基、羟基、氨基、氨甲基、马来酰亚胺、羧基、巯基、丙醛、琥珀酰亚胺碳酸酯、琥珀酰亚胺乙酸酯、琥珀酰亚胺丙酸酯、琥珀酰亚胺琥珀酸酯、琥珀酰亚胺、二硫吡啶基、丙酸基、醛基、正吡啶二硫基、巯酯基、丙烯酸基、叠氮基、戊二酸基、酰肼基、炔基、对硝基苯碳酸酯基、异氰酸基、邻二硫吡啶基、硅烷基、羧甲基。
优选的,所述的Z为甲酯、乙酯、叔丁酯、叠氮基、缩醛基或苄氧基。
E1为多缩乙二醇基团,其结构为:(CH2CH2O)j,j为0-100的整数。
在本发明的一个具体实施方式中,所述的步骤(1)可以为:在一个端基已修饰的多缩乙二醇衍生物中加入按体积比1-100倍,优选为1-20倍,更有选为1-10倍的二氯甲烷(DCM)及按摩尔比1-5倍,优选为1-3倍的三乙胺(TEA),然后将按摩尔比1-3倍,优选为1-2倍的甲磺酰氯(MsCl)或对甲苯磺酰氯溶于按体积比1-30倍,优选为1-20倍,更有选为1-10倍的二氯甲烷里,并滴加至反应瓶中,室温反应1-48小时,优选为2-10小时后,将反应液用水洗涤1-3次,蒸干溶剂得到产物,即端基已修饰的多缩乙二醇衍生物的磺酰化产物。
进一步,上述步骤后,还包括:在一个端基已修饰的多缩乙二醇衍生物的磺酸酯衍生物中加入按体积比1-100倍,优选为1-20倍,更有选为1-10倍的二甲基甲酰胺(DMF)及1-5倍摩尔比的卤化锂,然后加热至30-80℃,反应2-48小时,优选为2-10小时,反应完毕后,有机相用盐水洗涤1-3次,粗品蒸干后,柱层析纯化得到产物,即得一个端基已 修饰的多缩乙二醇的卤化产物。优选的,所述的卤化锂为溴化锂,所述的卤化产物为溴化产物。
在本发明的一个优选的实施方式中,所述的步骤(1)将端基已修饰的多缩乙二醇衍生物进行磺酸酯化的反应如下所示:
其中,所述的R为甲基或对甲苯基。
本发明所述Y型多缩乙二醇衍生物的制备方法步骤(2)中,一端端基为氨基的多缩乙二醇衍生物结构式可表示为:NH2-(CH2)m-E2-X-Z;
其中:
m为0-30的整数;
E2为多缩乙二醇基团,其结构可以表示为:(CH2CH2O)j,其中,j为从0-100的整数;
X为连接基团,选自:-(CH2)i-、-(CH2)iNH-、-(CH2)iOCOO-、-(CH2)iOCONH-、-(CH2)iNHCONH-、-OC(CH2)iCOO-、-(CH2)iCOO-、-(CH2)iCONH-,i为从0-10的整数,
Z为本发明所述的式(Ⅰ)结构中Y所定义的活性端基或者Z选自以下基团:甲酯、乙酯、叔丁酯、缩醛基、苄氧基。
所述Y为:C1-C6的烷氧基、羟基、氨基、氨甲基、马来酰亚胺、羧基、巯基、丙醛、琥珀酰亚胺碳酸酯、琥珀酰亚胺乙酸酯、琥珀酰亚胺丙酸酯、琥珀酰亚胺琥珀酸酯、琥珀酰亚胺、二硫吡啶基、丙酸基、醛基、正吡啶二硫基、巯酯基、丙烯酸基、叠氮基、戊二酸基、酰肼基、炔基、对硝基苯碳酸酯基、异氰酸基、邻二硫吡啶基、硅烷基、羧甲基。
优选的,所述的Z为甲酯、乙酯、叔丁酯、叠氮基、缩醛基或苄氧基。
在本发明一个具体实施方式中,所述Y型多缩乙二醇衍生物的制备方法步骤(2)包括:将一端端基为氨基的多缩乙二醇衍生物中加入按摩尔比为1-5倍的步骤(1)的产物及按体积比为1-100倍,优选为1-20倍,更有选为1-10倍的四氢呋喃(THF)或N,N-二甲基甲酰胺,在70-90℃下反应5-72小时,优选为2-10小时,蒸干溶剂后,粗品进行柱层析纯化得到产物。
在本发明的一个优选的实施方式中,所述Y型多缩乙二醇衍生物的制备方法步骤(2)的反应如下所示:
或者,
在本发明的另一个优选的实施方式中,所述Y型多缩乙二醇衍生物的制备方法步骤(2)的反应如下所示:
或者,
本发明所述制备方法步骤(3)可以根据所要制备得到所述的Y型多缩乙二醇衍生物中A或B的不同基团进行。根据Y型多缩乙二醇衍生物所应用的领域及方式,有时需要对所合成的Y型衍生物进行改造,即将Y型衍生物的1-3个端基改造成所需的活性基团。这种改造可在第一步之前进行:例如先合成一个端基为被保护的活性基团的多缩乙二醇衍生物,将其甲磺酸化,Y型化后脱保护,得到两个端基为活性基团的Y型衍生物。也可先合成Y型衍生物,然后针对A或B端基进行改造,得到一个端基为活性基团的Y型衍生物。也可以同时应用上述两种方式,得到三个端基都被改造的Y型衍生物。具体的改造方式有很多种,可以采用本领域可很容易获得其相关的合成方法,如羧基化及接下来的NHS化、胺基化、醛基化、巯基化、马来酰亚胺化、丙烯酸化等。
本发明所述的Y型多缩乙二醇化合物具有分子量均一,药物负载量高的优点,并且采用本发明所述的Y型多缩乙二醇化合物与难溶性药物分子连接后,与药物分子以及药物分子的直链聚乙二醇衍生物相比能够明显的改善水溶性。另外,本发明所述的Y型多缩乙二醇化合物的制备方法步骤简单,条件温和,易于工业化操作。
附图说明
图1是Y型多缩乙二醇衍生物(mEG3)2N-C3H6-OH的合成路线图。
图2是Y型多缩乙二醇衍生物(mEG5)2N-C3H6-OH的合成路线图。
图3是Y型多缩乙二醇衍生物(HO-EG4)2N-C3H6-OH的合成路线图。
图4是Y型多缩乙二醇衍生物(NH2-EG4)2N-C3H6-OH的合成路线图。
图5是Y型多缩乙二醇衍生物(HOOC-EG4)2N-C2H4-NH2的合成路线图。
图6是Y型多缩乙二醇衍生物(mEG3)2N-EG4-OH的合成路线图。
图7是胆固醇及胆固醇衍生物在水溶液中的分散性对比试验结果(由左至右分别为(mEG3)2N-C3H6-OH的胆固醇衍生物,mEG7-OH的胆固醇衍生物及胆固醇相同浓度的水溶液)。
具体实施方式
实施例1:(mEG3)2N-C3H6-OH的合成
其合成路线如图1所示。
1、mEG3-OMs的合成
在mEG3-OH(32mL,200mmol)中加入TEA(32mL,230mmol)及150mL DCM,置于冰水浴中的反应瓶内。用DCM(50mL)溶解MsCl(17.5mL,220mmol),溶解完全后,滴加至冰水浴中的反应瓶内。室温反应3小时。通过薄层色谱(TLC)检测反应完全。用水(150mL)洗三遍。有机相用无水硫酸钠干燥,过滤除去硫酸钠。浓缩得到产物约52g。
2、Y型小分子PEG的合成
氨基丙醇(3.1g,41.3mmol)中加入由上述步骤1制得的mEG3-OMs(21.6g,89.3mmol)及THF(150mL)。加热回流过夜后,倒出上层液,蒸干得粗品。柱纯化(取250g硅胶,流动相为MeOH/DCM体系,MeOH/DCM=3-7%),得到产品,产率:2.5g(16%)。
NMR(CDCl3)δ:3.5-3.8(m,22H,OCH2),3.37(s,6H,CH3O),2.7-2.8(m,6H,N(CH2)3,1.6-1.7(m,2H,NCH2CH2CH2OH);ESI-MS:368.3(M+H)+,390.2(M+Na)+。
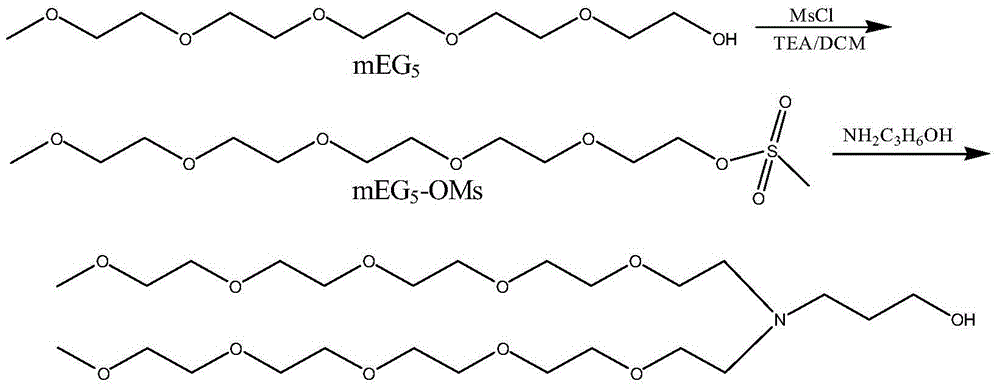
实施例2:(mEG5)2N-C3H6-OH的合成
其合成路线如图2所示。
1、mEG5-OMs的合成
在mEG5-OH(10.08g,40mmol)中加入TEA(6.86mL,48mmol)及DCM(60mL),置于冰水浴中的反应瓶内。用DCM(40mL)溶解MsCl(3.56mL,46mmol)。滴加至冰水浴中的反应瓶内。室温反应3小时。TLC检测反应完全。用水(150mL)洗三遍。有机相用无水硫酸钠干燥,过滤除去硫酸钠。浓缩得到产物约10g。
2、Y型小分子PEG的合成
在上述步骤1制得的mEG5-OMs(4g,12.1mmol)中加入氨基丙醇(0.45g,6.06mmol)、TEA(1mL)及THF(100mL),加热回流过夜后,倒出上层液。蒸干得粗品。柱纯化(流动相为MeOH/DCM体系,MeOH/DCM=3-7%),得到产品,产率:0.3g(9.1%)。
NMR(CDCl3)δ:3.5-3.8(m,38H,OCH2),3.37(s,6H,CH3O),2.7-2.8(m,6H,N(CH2)3,1.6-1.7(m,2H,NCH2CH2CH2OH);ESI-MS:544.4(M+H)+,566.3(M+Na)+。
实施例3:(HO-EG4)2-C3H6-OH的合成
其合成路线如图3所示。
在HO-EG4-Br(21g,81.7mmol)中加入氨基丙醇(3.06g,40.9mmol)及THF(150mL)加热回流过夜,冷却后蒸干溶剂得粗品。柱纯化(流动相为MeOH/DCM体系,MeOH/DCM=0-10%),得到产品,产率:2.3g(13.1%)。
NMR(CDCl3,盐酸盐)δ:3.4-3.9(m,28H,-OCH2-),3.3(m,2H,-CH2CH2CH2OH),3.1-3.2(m,6H,-NCH2-),2.0(m,2H,-CH2CH2CH2OH);ESI-MS:428.4(M+H)+。
实施例4:(NH2-EG4)2N-C3H6-OH的合成
其合成路线如图4所示。
1、N3-EG4-OH的合成
在反应瓶中加入EG4(36mL,210mmol)、DCM(100mL)及TEA(25mL)。冰水浴。滴加含MsCl(5.81mL,75mmol)的DCM溶液(100mL)。室温反应4小时。加入水(100mL)洗涤一次,蒸干得到粗品。
将上一步粗品中加入95%乙醇(150mL)及叠氮钠(6.5g,100mmol)。室温回流16小时。过滤除去固体。蒸干溶液。加入DCM(150mL)并用水(100mL)洗涤三次。DCM相蒸干得到粗品14g。柱纯化(取250g硅胶,流动相为PE/EA体系,PE/EA=50-0%),得到产品,产率:12.3g(26.7%)。
2、N3-EG4-OMs的合成
向上述步骤1制得的N3-EG4-OH(11g,50.2mmol)中加入TEA(8.6mL,60.3mmol,1.2 eq)及DCM(150mL)。冰水浴。用DCM(50mL)溶解MsCl(4.5mL,57.8mmol,1.15eq)。滴加至冰水浴中的反应瓶内。室温反应过夜。TLC检测反应完全。用水(100mL)洗三遍。有机相用无水硫酸钠干燥,过滤除去硫酸钠。浓缩得到产物。
3、(N3-EG4)2N-C3H6-OH的合成
向上述步骤2制得的N3-EG4-OMs(14.5g,48.8mmol)中加入氨基丙醇(1.86mL,24.4mmol)及DMF(130mL),加热回流过夜后。蒸干得粗品。柱纯化(流动相为MeOH/DCM体系,MeOH/DCM=3-7%),得到产品,产率:1.2g(10.3%)。
4、(NH2-EG4)2N-C3H6-OH的合成
向上述步骤3制得的(N3-EG4)2N-OH(400mg,0.84mmol)中加入DMF(20mL)及三苯基膦(615mg,2.35mmol,1.4eq*2),室温反应过夜后,加入水(0.1mL),反应过夜。蒸干DMF,加入水(50mL),依次用甲苯(40mL)洗涤2次及DCM(30mL)洗涤2次,蒸干水分得到产物,产率:300mg(84.0%)。
NMR(D2O)δ:3.5-3.8(m,38H,OCH2),2.4-2.8(m,10H,N(CH2)3及NH2CH2,1.6-1.7(m,2H,NCH2CH2CH2OH);ESI-MS:448.4(M+Na)+。
实施例5:(HOOC-EG4)2N-C2H4-NH2的合成
其合成路线如图5所示。
1、MsO-EG4-OtBu的合成
在HO-EG4-tBu(39.4g,0.122mol)中加入DCM(350mL)及TEA(22.7mL,0.159mol,1.3eq)。冰水浴下加入MsCl(11.4mL,0.146mol,1.2eq)的DCM(150mL)溶液。室温反应过夜。依次用水(200mL)洗涤3次,无水硫酸钠干燥,过滤,蒸除溶剂得产物。
2、(tBuO-EG4)2N-C2H4-NH-Boc的合成
向上述步骤1制得的tBu-EG4-OMs(20g,50mmol)中加入NH2-C2H4-NH-Boc(4g,25mmol)及THF(200mL),室温搅拌过夜。浓缩得粗品。柱纯化(MeOH/DCM=0-10%)得到产品,产率:2.3g(12.0%)。
3、(HOOC-EG4)2N-C2H4-NH2的合成
向上述步骤2制得的(tBuO-EG4)2N-C2H6-NH-Boc(2.3g,3.0mmol)中加入DCM(20mL)及三氟乙酸(TFA)(8mL),室温反应过夜,蒸干溶剂,蒸干溶剂得到产品,产率:2.2g(94.0%)。NMR(CDCl3)δ:2.6-2.7(t,4H,CH2COO);3.5-3.8(m,40H,其他氢);ESI-MS:557.4(M+H)+,595.3(M+Na)+。
实施例6:(mEG3)2N-EG4-OH的合成
1、NH2-EG4-OH的合成
在N3-EG4-OH(9.2g,42.0mmol)中加入THF(50mL)。冰水浴。滴加含三苯基膦(13.21g,50.4mmol,1.2eq)的THF(75mL)。室温反应24小时。加入水(1965uL,109.2mmol,2.6eq)。室温反应过夜。蒸干溶剂,加入水(150mL)。用甲苯(150mL)及DCM(100mL)洗涤。蒸干水相得产品8g。
2、(mEG3)2N-EG4-OH的合成
向EG3-OMs(10.8g,44.6mmol)中加入由步骤1制得的NH2-EG4-OH(4.3g,22.3mmol)及THF(150mL),回流搅拌过夜。蒸干溶剂得到粗品。柱纯化(流动相为MeOH/DCM体系,MeOH/DCM=3-7%),得到产品2.1g(19.4%)。NMR(CDCl3)δ:3.5-3.8(m,34H,OCH2),3.37(s,6H,CH3O),2.7-2.8(m,6H,N(CH2)3;ESI-MS:486.4(M+H)+,508.4(M+Na)+。
实施例7:(mEG3)2N-C3H6-OH的胆固醇衍生物合成
取实施例1制备得到的(mEG3)2N-C3H6-OH(2g,5.45mmol)中加入DCM(20mL)及TEA(0.905mL,6.53mmol),然后将胆固醇氯甲酸酯(2.57g,5.72mmol)溶于DCM(30mL)中,滴加至反应瓶中。室温搅拌过夜。TLC检测反应完全,水洗一遍,旋干得粗品。柱纯化(流动相为MeOH/DCM体系,MeOH/DCM=0-7%),得到产品1.4g(32.4%)。
NMR(CDCl3)δ:5.55-5.45(m,1H),4.65-4.50(m,1H),4.1-4.0(m,2H),3.5-3.8(m,20H),3.37(s,6H),2.7-2.8(m,6H),2.50-0.80(m,42H),0.65-0.60(m,3H)。
实施例8:mEG7-OH的胆固醇衍生物合成
取市售获得的mEG7-OH,按照实施例7相同条件制备mEG7-OH的胆固醇衍生物。
NMR(CDCl3)δ:5.55-5.45(m,1H),4.65-4.50(m,1H),4.1-4.0(m,2H),3.5-3.8(m,26H),3.37(s,3H),2.50-0.80(m,40H),0.65-0.60(m,3H)。
实施例9:胆固醇及胆固醇衍生物在水中的分散性比较试验
分别将实施例7制备获得的(mEG3)2N-C3H6-OH的胆固醇衍生物,实施例8制备获得的mEG7-OH的胆固醇衍生物及胆固醇各20mg放置于50mL容量瓶中,加水至刻度。将其置于20℃水浴中,每隔5min剧烈震摇30min,30min后,两种胆固醇衍生物与胆固醇在水中的分散状况如图7所示,从左至右依次为(mEG3)2N-C3H6-OH的胆固醇衍生物,mEG7-OH的胆固醇衍生物及胆固醇。肉眼观察结果,(mEG3)2N-C3H6-OH的胆固醇衍生物分散液呈半透明的淡蓝色,可清晰看到容量瓶背后的字迹;mEG7-OH的胆固醇衍生物则是白色乳浊液,而胆固醇完全不溶解。
实施例10:胆固醇衍生物在水中溶解度比较试验
按《中国药典》方法测定胆固醇衍生物在水中溶解度:
将实施例7制备得到的(mEG3)2N-C3H6-OH的胆固醇衍生物21.3mg分散于50mL水中,从中取出5mL,加入1.5mL水,将其置于20℃水浴中,每隔5min剧烈震摇30min,30min后,又加入1.5mL水,同样置于20℃水浴中以相同操作进行溶解,直至加入9mL水,溶液澄清。
将实施例8制备得到的mEG7的胆固醇衍生物24.2mg分散于50mL水中,从中取出5mL,加入5mL水,将其置于20℃水浴中,每隔5min剧烈震摇30min,30min后,又加入5mL水,同样置于20℃水浴中以相同操作进行溶解,直至加入25mL水。再从稀释溶液中取出5mL溶液,同样操作直至加入10mL水,溶液澄清。
由结果分析可知,(mEG3)2N-C3H6-OH的胆固醇衍生物溶解度为15.2mg/100g,是mEG7-OH的胆固醇衍生物的溶解度4.61mg/100g的3.3倍,是胆固醇溶解度(<0.2mg/100g)的76倍。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种新型罗汉果醇衍生物单体的制作方法
- 通过预先占位调控邻位碳硼烷衍生物结构的方法
- Schiglautone A衍生物的组合物用于制备抗鼻炎药物的制作方法
- Schiglautone A衍生物的组合物用于制备防治肾纤维化药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗心衰药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗菌药物的制作方法
- Schiglautone A 衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid的衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid的衍生物的组合物用于制备防治胰腺纤维化药物的制作方法
- 毛栲利素衍生物的制备方法及其抗菌活性的新用图
- 6-o-脱甲基侧厚壳桂碱衍生物及其制备和抗病毒活性应用及抗癌活性的制作方法
- 一种聚乙二醇中吡啶衍生物的合成方法
- 棉酚衍生物和它们的制备,在农药上的应用及抗癌活性的制作方法
- 用于治疗或预防神经精神病疾病的含有黄酮-6-c-葡萄糖衍生物作为活性成分的药物组合物的制作方法
- 具有抗癌活性的6-氧代-1,6-二氢-哒嗪衍生物与吉非替尼的组合的制作方法
- 2,3′-脱水-2′-脱氧-5-氟尿苷衍生物与细胞毒活性、制备方法及应用
- 2,3′-脱水-2′-脱氧-5-氟尿苷衍生物与细胞毒活性、制备方法及应用
- 具有抗肿瘤活性的哌啶酮衍生物及其制备方法
- 一种支化聚乙二醇的脂质衍生物及其组成的脂质膜结构体的制作方法
- 一种新型罗汉果醇衍生物单体的制作方法
- 通过预先占位调控邻位碳硼烷衍生物结构的方法
- Schiglautone A衍生物的组合物用于制备抗鼻炎药物的制作方法
- Schiglautone A衍生物的组合物用于制备防治肾纤维化药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗心衰药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid衍生物的组合物用于制备抗菌药物的制作方法
- Schiglautone A 衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid的衍生物的组合物用于制备抗慢性阻塞性肺病药物的制作方法
- Artalbic acid的衍生物的组合物用于制备防治胰腺纤维化药物的制作方法