苯并吡唑联吡啶类化合物、包含此类化合物的药物组合物及其用途的制作方法
本发明涉及药物化学和药物治疗学领域,具体涉及作为受体酪氨酸激酶EGFR,Her2,VEGFR,FGFR等多靶点抑制剂的苯并吡唑联吡啶类化合物、其制备方法、包含此类化合物的药物组合物及用途。
背景技术:
长期以来,恶性肿瘤的治疗是一个世界性的难题,其特点是细胞或变异细胞异常增殖。以往对肿瘤的治疗是通过发现肿瘤并破坏来实现,随着对细胞信号传导途径的不断研究,人们对肿瘤细胞内部的肿瘤基因及抗肿瘤基因的作用了解得越来越深入,使得针对肿瘤的特异性分子靶点设计的新的抗肿瘤药成为可能。
酪氨酸激酶(Tyrosine Kinases,TKs)是信号传递过程中的重要因子,通过催化三磷酸腺苷(ATP)的磷酸基转移到许多重要蛋白质的酪氨酸残基上,使酚羟基磷酸化,从而传递信号。根据TKs是否存在于细胞膜上,可将其分为受体酪氨酸激酶(Receptor Tyrosine Kinase,RTK)和非受体酪氨酸激酶(Nonreceptor Tyrosine Kinase,NTK)。受体酪氨酸激酶主要包括血小板生长因子受体(PDGFR)、成纤维细胞生长因子受体(FGFR)、表皮生长因子受体(EGFR)和血管内皮生长因子受体(VEGFR)等,他们通常具有一个细胞外结构域、一个跨膜区以及一个细胞内激酶域;非受体酪氨酸激酶包括Src和Jak等家族。蛋白酪氨酸激酶PTK在细胞内的信号转导中起着十分重要的作用,它参与正常细胞的调节、信号传递和发育,也与肿瘤细胞的增殖、分化、迁移和凋亡密切相关。酪氨酸激酶功能的失调,会导致其下游信号途径激活,引起细胞增殖调节紊乱,最终导致肿瘤形成。
目前抑制酪氨酸激酶主要通过两种途径,即单克隆抗体和小分子酪氨酸激酶抑制剂(Tyrosine Kinases Inhibitors,TKIs)单克隆抗体可与细胞外受体结合,阻断其功能。小分子抑制剂主要表现为与ATP结合位点发生作用,抑 制细胞内蛋白激酶的活化。小分子酪氨酸激酶抑制剂包括单靶点和多靶点酪氨酸激酶抑制剂。后者通过抑制多种传导通路达到抗肿瘤作用,相对于前者,其在疗效及患者耐受性方面都有很大优势。因此,以酪氨酸激酶为靶点进行的药物研发成为国际上抗肿瘤药物研究的热点。
多靶点酪氨酸激酶是一个富有前景的抗肿瘤药物研究的靶标。目前,多靶点酪氨酸激酶小分子抑制剂的结构类型有限,仍然无法克服抑制剂产生耐药性及毒性等问,开发新型母核的抑制剂就显得十分重要。
技术实现要素:
本发明的一个目的在于提供一种通式(I)所示的苯并吡唑联吡啶类化合物、其可药用的盐、对映异构体、非对映异构体或外消旋体。
本发明的另一个目的在于提供一种上述通式(I)所示化合物的制备方法。
本发明的再一个目的在于提供一种包含治疗有效量的一种或多种上述通式(I)所示化合物或其可药用的盐的药物组合物。
本发明的又一个目的在于提供上述通式(I)所示化合物在制备用于治疗酪氨酸激酶EGFR,Her2,VEGFR,FGFR等信号转导通路相关的细胞增生疾病的药物中的用途。
本发明的又一个目的在于提供上述通式(I)所示化合物在用于治疗酪氨酸激酶EGFR,Her2,VEGFR,FGFR等信号转导通路相关的细胞增生疾病中作为酪氨酸激酶抑制剂的用途。
本发明的又一个目的在于提供上述通式(I)所示化合物在抑制酪氨酸激酶中的用途。
本发明的又一个目的在于提供一种治疗酪氨酸激酶EGFR,VEGFR信号转导通路相关的细胞增生疾病的方法,其特征在于,向受试者施用治疗有效量的一种或多种上述通式(I)所示化合物或其可药用的盐。
本发明提供了一种通式(I)所示化合物、其可药用的盐、对映异构体、非对映异构体或外消旋体,
其中:
R1为C1-C12直链或支链的烷基、C2-C12直链或支链的不饱和烃基、取代或未取代的C3-C12环烃基、取代或未取代的C3-C12杂环基、取代或未取代的C6-C10芳基、或者取代或未取代的C5-C12杂芳基,所述杂环基或杂芳基含有1~4个选自氧、硫和氮中的杂原子;
其中,R1中所述取代的取代基为卤素、C1-C12直链或支链的烷基、C1-C12直链或支链的烷氧基、C2-C12直链或支链的不饱和烃基、C3-C12环烃基、氰基、硝基、氨基、羟基、三氟甲基、三氟甲氧基、羧基、巯基、苯基、磺酰胺基、萘基、联苯基、C5-C12杂芳基或C3-C12杂环基,所述杂环基或杂芳基含有1~4个选自氧、硫和氮中的杂原子;
优选地,R1为取代或未取代的C6-C10芳基、或者取代或未取代的C5-C12杂芳基,所述杂芳基含有1~4个选自氧、硫和氮中的杂原子;
其中,R1中所述的取代基为卤素、C1-C12直链或支链的烷基、C1-C12直链或支链的烷氧基、C2-C12直链或支链的不饱和烃基、C3-C12环烃基、氰基、硝基、氨基、羟基、三氟甲基、三氟甲氧基、羧基、磺酰胺基、巯基;
更优选地,R1为:
其中,R4为1-5个取代基,其各自独立地为氢、卤素、C1-C12直链或支链的烷基、C2-C12直链或支链的不饱和烃基、C3-C12环烃基、氰基、硝基、氨基、羟基、三氟甲基、三氟甲氧基、羧基、巯基;
优选地,R4为1-5个取代基,其各自独立地为氢或卤素;
在通式(I)中,R2、R3各自独立地为氢、卤素、氰基、硝基、氨基、羟基、羟甲基、甲氧基、三氟甲基、三氟甲氧基、羧基、C1–C6直链或支链的烷基、 C2-C6直链或支链的不饱和烃基、C1–C6直链或支链的烷氧基、C1–C6直链或支链的烷酰基或C1–C6直链或支链的烷氨基;
优选地,R2、R3各自独立地为氢、卤素、氨基、硝基、羟基、甲氧基、三氟甲基、三氟甲氧基、C1–C6直链或支链的烷基、C2-C6直链或支链的不饱和烃基、C1–C6直链或支链的烷氧基、C1–C6直链或支链的烷酰基或C1–C6直链或支链的烷氨基;
更优选地,R2、R3为氢、卤素、氨基、硝基、羟基、甲氧基、三氟甲基、三氟甲氧基、C1–C6直链或支链的烷基、C2-C6直链或支链的不饱和烃基、C1–C6直链或支链的烷氧基;
更优选地,R2、R3为氢、氟、氯、氨基、三氟甲基、C1–C6直链或支链的烷基;
为取代或未取代的C6-C12芳基、取代或未取代的C3-C12杂环基,取代或未取代的C5-C12杂芳基,所述杂环基或杂芳基含有1-4个选自氧、硫和氮的杂原子,其中所述取代的芳基、杂环基或杂芳基包括1~5个取代基,该取代基各自独立地为卤素、C1-C12直链或支链的烷基、C1–C6直链或支链的烷氧基、C2-C12直链或支链的不饱和烃基、C3-C12环烃基、羟甲基、氰基、硝基、氨基、羟基、羟甲基、三氟甲基、三氟甲氧基、羧基、巯基、COOR5、CONR5R6、NR5SO2R6、NR5R6、NR5COR6,或者任意两个相邻的取代基连接成环;
其中,R5、R6各自独立地为氢、C1-C12直链或支链的烷基、C2-C12直链或支链的不饱和烃基、C3-C12环烃基、C5-C12芳基、取代或未取代的C5-C12杂芳基、取代或未取代的C3-C12杂环基;所述杂环基或杂芳基含有1-4个选自氧、硫和氮的杂原子,并且取代的杂环基或杂芳基含有一个或多个选自卤素、C1-C12直链或支链的烷基、C2-C12直链或支链的不饱和烃基、C1-C6直链或支链的烷氧基、C3-C12环烃基、C3-C12杂环基、氰基、硝基、氨基、羟基、羟甲基、三氟甲基、三氟甲氧基、羧基、巯基中的取代基,或者任意两个相邻的取代基连接成环;
优选地,为取代或未取代的如下基团:
其中,所述取代的上述基团包括1~3个取代基,该取代基各自独立地为卤素、C1-C12直链或支链的烷基、C1-C6直链或支链的烷氧基、羟甲基、氰基、氨基、羟基、三氟甲基、三氟甲氧基、CONR5R6、NR5SO2R6、NR5R6、NR5COR6,或者任意两个相邻的取代基连接成环;
在本发明更优选的实施方案中,本发明的通式I的化合物优选为如下具体化合物:
化合物编号和结构如下表1所示:
表1
本发明的化合物可能具有不对称中心、手性轴和手性平面并且可以以对映异构体、非对映异构体、外消旋体及其混合物的形式存在。
本发明提供了通式(I)化合物的可药用的盐,具体地为通式(I)化合物与无机酸或有机酸反应形成的常规的无毒盐。例如,常规的无毒盐可通过通式(I)化合物与无机酸或有机酸反应制得,所述无机酸包括盐酸、氢溴酸、硫酸、硝酸、胺基磺酸和磷酸等,以及所述有机酸包括柠檬酸、酒石酸、乳酸、丙酮酸、乙酸、苯磺酸、对甲苯磺酸、甲磺酸、萘磺酸、乙磺酸、萘二磺酸、马来酸、苹果酸、丙二酸、富马酸、琥珀酸、丙酸、草酸、三氟乙酸、硬酯酸、扑酸、羟基马来酸、苯乙酸、苯甲酸、水杨酸、谷氨酸、抗坏血酸、对胺基苯磺酸、2-乙酰氧基苯甲酸和羟乙磺酸等;或者通式(I)化合物与丙酸、草酸、丙二酸、琥珀酸、富马酸、马来酸、乳酸、苹果酸、酒石酸、柠檬酸、 天冬氨酸或谷氨酸形成酯后再与无机碱形成的钠盐、钾盐、钙盐、铝盐或铵盐;或者通式(I)化合物与有机碱形成的甲胺盐、乙胺盐或乙醇胺盐;或者通式(I)化合物与赖氨酸、精氨酸、鸟氨酸形成酯后再与盐酸、氢溴酸、氢氟酸、硫酸、硝酸、磷酸形成的对应的无机酸盐或与甲酸、乙酸、苦味酸、甲磺酸和乙磺酸形成的对应的有机酸盐。
本发明提供了一种通式(I)表示的化合物的制备方法,该制备方法按照如下步骤,包括:
其中,R1、R2、R3和的定义如前述,
步骤a:将2-氯-3溴-5氨基吡啶分散在溶剂中。在冰浴条件下,再依次加入亚硝酸钠和碘化钾,待反应完全后,加入NaOH水溶液中和,得化合物Ia,所述溶剂为盐酸水溶液;
步骤b:将化合物Ia分散在溶剂中,加入氨基醇类衍生物、磷酸钾、乙二醇和碘化亚铜,加热到90度反应24小时,得化合物Ib,所述溶剂为异丙醇;
步骤c:将苯并吡唑类衍生物溶解在溶剂中,加入Boc酸酐、DMAP和三乙胺在室温下反应8小时,得化合物Ic,所述溶剂为二氯甲烷;
步骤d:将化合物Ic溶解在溶剂中,加入联频那醇硼酸酯、Pd(dppf)2Cl2和醋酸钾,加热到80度反应6小时,得化合物Id,所述溶剂为二氧六环;
步骤e:将化合物Ib和Id溶解在溶剂中,加入Pd(dppf)2Cl2和2M的碳酸钠水溶液,加热到90度反应24小时,所述溶剂为二氧六环;
步骤f:将步骤e所得溶液过滤、萃取、悬干后溶解于溶剂中,加入三氟乙酸,室温反应过夜得化合物Ie,所述溶剂为二氯甲烷;
步骤g:将化合物Ie和A环硼酸衍生物溶解在溶剂中,加入Pd(dppf)2Cl2和磷酸钾水溶液,微波加热到120度反应2小时,得化合物If,所述溶剂为二氧六环。
本发明提供了上述通式(I)所示化合物在制备用于治疗酪氨酸激酶EGFR,Her2,VEGFR,FGFR等信号转导通路相关的细胞增生疾病的药物中的用途;
本发明还提供了上述通式(I)所示化合物在治疗酪氨酸激酶EGFR,Her2,VEGFR,FGFR等信号转导通路相关的细胞增生疾病中的用途;
本发明的还提供一种治疗酪氨酸激酶EGFR,Her2,VEGFR,FGFR等信号转导通路相关的细胞增生疾病的方法,其特征在于,向受试者施用治疗有效量的一种或多种上述通式(I)所示化合物或其可药用的盐;
所述酪氨酸激酶EGFR,Her2,VEGFR,FGFR等信号转导通路相关的细胞增生疾病例如为癌症、超常增生、再狭窄、免疫病症和炎症;
本发明所述癌症包括但不限于,组织细胞性淋巴瘤、卵巢癌、头颈磷状上皮细胞癌、胃癌、乳腺癌、儿童肝细胞癌、结肠直肠癌、宫颈癌、肺癌、肉瘤、鼻咽癌、胰腺癌、成胶质细胞癌、前列腺癌、小细胞肺癌、非小细胞肺癌、多发性骨髓瘤、甲状腺癌、睾丸癌、宫颈癌、子宫内膜癌、食道癌、白血病、肾细胞癌、膀胱癌、肝癌和星形细胞瘤等;更优选地用于治疗如下癌症:头颈磷状上皮细胞癌、组织细胞性淋巴瘤、肺腺癌、小细胞肺癌、非小细胞肺癌、胰腺癌、乳头状肾细胞癌、肝癌、胃癌、结肠癌、多发性骨髓瘤和成胶质细胞瘤;
本发明的化合物和组合物还可用于治疗、预防或调控癌细胞和癌症的转移瘤,特别是用于预防或调控卵巢癌、儿童肝细胞癌、转移性的头颈磷状上皮细胞癌、胃癌、乳腺癌、结肠直肠癌、宫颈癌、肺癌、鼻咽癌、胰腺癌、成胶质细胞瘤和肉瘤的转移瘤。
本发明还提供了上述通式(I)所示化合物在抑制酪氨酸激酶中的用途。
本发明还提供了一种药物组合物,其含有治疗有效量的上述通式(Ⅰ)的化合物或其可药用的盐,以及含有一种或多种可药用的载体;该药用组合物还可以进一步包含气味剂、香味剂等。
本发明所述的药物组合物优选含有重量比为1~99%的活性成份,其优选的比例是,通式(I)化合物作为活性成分占总重量比65%~99%,其余部分为药学可接受的载体、稀释液或溶液或盐溶液。
本发明所述的化合物和药物组合物可以是多种形式,如片剂、胶囊、粉剂、糖浆、溶液状、悬浮液和气雾剂等,并可以存在于适宜的固体或液体的载体或稀释液中和适宜的用于注射或滴注的消毒器具中。
本发明的药物组合物的各种剂型可按照药学领域的常规制备方法制备。其制剂配方的单位剂量中包含0.05~200mg通式(I)化合物,优选地,制剂配方的单位剂量中包含0.1mg~100mg通式(I)化合物。
本发明的化合物和药物组合物可对哺乳动物临床使用,包括人和动物,可以通过口、鼻、皮肤、肺、或者胃肠道等给药途径进行给药。最优选为口服。最优选日剂量为0.01~200mg/kg体重,一次性服用,或0.01~100mg/kg 体重分次服用。不管用何种服用方法,个人的最佳剂量应依据具体的治疗而定。通常情况下是从小剂量开始,逐渐增加剂量一直到找到最适合的剂量。
附图说明
图1表示出了DCL16抑制了细胞水平EGFR激酶及下游信号分子的磷酸化。
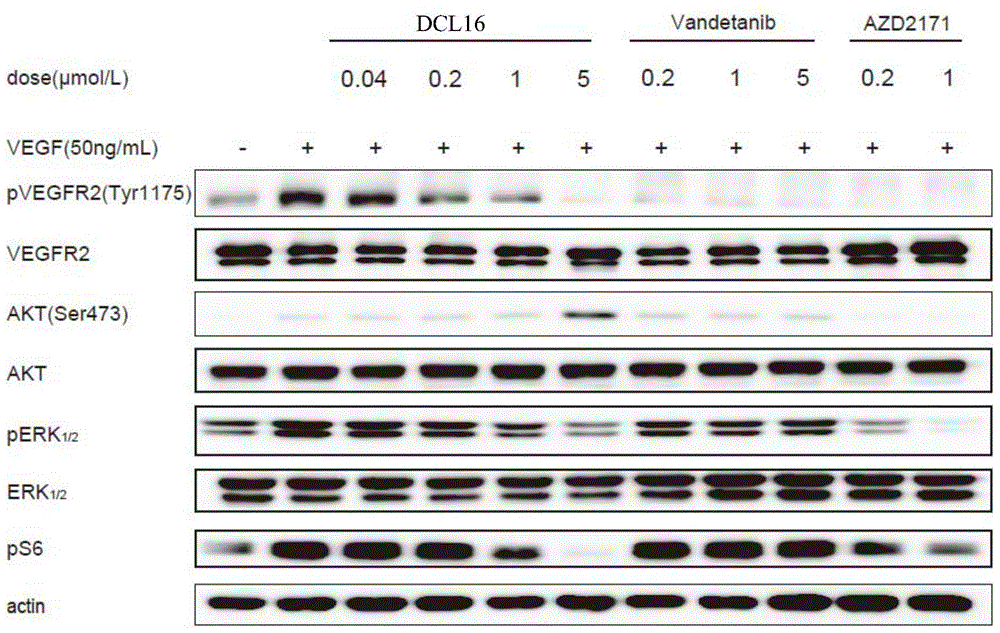
图2表示出了DCL16抑制了细胞水平VEGFR2激酶及下游信号分子的磷酸化。
具体实施方式
在以下的实施例中将进一步举例说明本发明。这些实施例仅用于说明本发明,但不以任何方式限制本发明。
本发明中用到的起始原料未经特别说明,均为商业购买。
流程1
实施例1
步骤1:制备2-氯-3溴-5碘吡啶
将2-氯3-溴-5碘吡啶(30.00g,144.61mmol),分散于200mL 5N盐酸水溶液中,在冰浴条件下,缓慢加入亚硝酸钠(14.96g,216.91mmol)水溶液,滴完5分钟后,在缓慢加入碘化钾(52.81,318.14mmol)水溶液,保持温度低于10度,滴完后再回到室温搅拌半小时后,再置于冰水浴中,加入5N NaOH水溶液中和至PH约为10-11,再用乙酸乙酯2×600mL萃取,有机相依次用300mL硫代硫酸钠水溶液、饱和食盐水洗涤,无水Na2SO4干燥,过滤,减压蒸除溶剂,得棕色油状物质,经柱层析(石油醚)分离纯化后,得固 体35.7g白色固体,产率78%。1H NMR(500MHz,CHCl3)δ9.21(s,1H),8.66(s,1H)。
步骤2:制备(N-2-氯3-溴5-吡啶)-2-氨基2-苯乙醇
将2-氯-3溴-5碘吡啶(2.5g,7.85mmol)溶解在15ml异丙醇中,再依次加入苯甘氨醇(1.62g,11.78mmol)、磷酸钾(5g,23.56mmol)、乙二醇(486mg,7.85mmol)和碘化亚铜(150mg,0.785mmol),用氩气置换三次后置于80度油浴中反应24h后,加入100ml二氯甲烷,有机相依次用3×30mL水、饱和食盐水洗涤,无水Na2SO4干燥,过滤,减压蒸除溶剂,得棕黑色油状物质,经柱层析((石油醚/乙酸乙酯8/1-4/1,v/v)分离纯化,得1.2g黄色固体,产率46%。1H NMR(500MHz,CHCl3)δ8.03(s,4H),7.64(s,4H),7.36(s,8H),7.32(s,6H),7.27(s,5H),4.60(s,2H),3.69(s,4H),3.45(d,J=9.1Hz,8H),1.16(s,4H)。
流程2
实施例2
步骤1:制备N-叔丁氧羰基-3-甲基5-溴苯并吡唑
将3-甲基5-溴苯并吡唑(1g,4.74mmol)溶解在15ml二氯甲烷中,依次加入Boc2O(1.55g,7.11mmol),三乙胺(1.03ml,7.11mmol)以及DMAP(57.88mg,0.47mmol),室温条件下反应4h后,加入40ml二氯甲烷,有机相依次用1N HCl(3×20mL)溶液、饱和食盐水洗涤,无水Na2SO4干燥,过滤,减压蒸除溶剂,,经柱层析((石油醚/乙酸乙酯20/1-10/1,v/v)分离纯化,得1.24g白色固体,产率84%。1H NMR(500MHz,CHCl3)δ8.21(s,1H),8.13(s,1H),7.61(s,1H),1.63(s,9H),1.57(s,3H)。
步骤2:制备N-叔丁氧羰基-3-甲基苯并吡唑5-频那醇硼酸酯
将N-叔丁氧羰基-3-甲基5-溴苯并吡唑(1.24g,3.98mmol)溶解在25ml二氧六环中,在氩气保护的条件下,依次加入联频哪醇硼酸酯(1.52g,5.98mmol),醋酸钾(977mg,9.96mmol)Pd(dppf)2Cl2(280mg,0.39mmol), 置于80度油浴反应过夜,所得溶液过滤后加入50ml乙酸乙酯,有机相依次用3×30mL水、饱和食盐水洗涤,无水Na2SO4干燥,过滤,减压蒸除溶剂,得黑色油状物质,经柱层析((石油醚/乙酸乙酯15/1-4/1,v/v)分离纯化,得1.16g白色固体,产率81%。1H NMR(500MHz,CHCl3)δ8.28(s,1H),1.62(s,9H),1.56(s,3H),1.13(s,12H)。
流程3
实施例3
步骤1:制备N-(2-氯-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇
将(N-2-氯-3-溴-5-吡啶)-2-氨基-2-苯乙醇(1g,3.05mmol)和N-叔丁氧羰基-3-甲基苯并吡唑5-频那醇硼酸酯(1.31g,3.66mmol)溶于20ml二氧六环溶液中,再加入2M碳酸钠溶液(3.82ml,7.63mmol)用氩气置换三次后,在氩气保护条件下加入Pd(dppf)2Cl2(223mg,0.3mmol),所得溶液过滤后加入50ml乙酸乙酯,有机相依次用3×30mL水、饱和食盐水洗涤,无水Na2SO4干燥,过滤,减压蒸除溶剂,得黑色油状物质。加入20ml二氯甲烷溶液和5ml三氟乙酸,室温反应过夜,有机相依次用3×30mL饱和碳酸氢钠、饱和食盐水洗涤,无水Na2SO4干燥,过滤,减压蒸除溶剂,得黑色油状物质,经柱层析(石油醚/乙酸乙酯4/1-1/1,v/v)分离纯化,得560mg淡黄色色固体,产率48%。1H NMR(500MHz,CHCl3)δ8.50(s,10H),8.09(s,10H),7.94(s,10H),7.72(d,J=23.0Hz,20H),7.39(dd,J=8.8,7.3Hz,2H),7.49–7.25(m,49H),4.63(s,9H),3.87–3.42(m,26H),3.44(s,7H),3.44(s,6H),1.57(s,29H),1.37(s,10H)。
流程4
实施例4
步骤1:制备N-(2-苯基-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇
将制备的N-(2-氯-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(100mg,0.27mmol)和苯硼酸(48mg,0.4mmol)加入2ml二氧六环中,再依次加入0.1ml水,碳酸钾(117mg,0.66mmol)和Pd(dppf)2Cl2(20mg,26μmol),用氩气置换三次空气后密封微波135度反应2小时。所得溶液加入20ml二氯甲烷,有机相依次用3×30mL水、饱和食盐水洗涤,无水Na2SO4干燥,过滤,减压蒸除溶剂,得黑色油状物质,经柱层析((二氯甲烷/甲醇50/1-20/1,v/v)分离纯化,得45mg白色固体,以41%产率得到化合物DCL01。1H NMR(500MHz,CHCl3)δ8.51(s,4H),8.19(s,8H),7.99(s,4H),7.74(d,J=15.0Hz,8H),7.65(s,8H),7.49(s,2H),7.37(d,J=8.5Hz,13H),7.32(s,6H),7.27(s,5H),4.95(s,2H),4.52(s,4H),3.69(s,3H),3.44(s,3H),1.57(s,12H),1.46(s,4H)。
实施例5
制备N-(2-(3-呋喃基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL02)
除了将苯硼酸换成3-呋喃硼酸之外,其他与实施例4相同方法制备化合物DCL02,产率42%。1H NMR(500MHz,CHCl3)δ8.51(s,4H),8.15(s,4H),7.94(s,4H),7.75(s,4H),7.63(s,4H),7.50(s,4H),7.44(s,5H),7.36(s,8H),7.32(s,6H),7.27(s,3H),5.73(s,4H),4.76(s,2H),4.08(s,4H),3.69(s,4H),3.44(s,4H),2.18(s,4H),1.57(s,12H)。
实施例6
制备N-(2-(3-噻吩基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL03)
除了将苯硼酸换成3-噻吩硼酸之外,其他与实施例4相同方法制备化合物DCL03,产率40%。1H NMR(500MHz,CHCl3)δ8.51(s,4H),8.14(s,4H), 7.98(s,4H),7.81(d,J=60.0Hz,8H),7.64(s,4H),7.40(d,J=5.7Hz,2H),7.39–7.30(m,17H),7.27(s,5H),7.04(s,4H),4.73(s,2H),3.98(s,4H),3.69(s,4H),3.44(s,4H),1.57(s,12H),1.49(s,4H)。
实施例7
制备N-(2-(2-吡咯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL04)
除了将苯硼酸换成3-噻吩硼酸之外,其他与实施例4相同方法制备化合物DCL04,产率38%.1H NMR(500MHz,CHCl3)δ8.68(s,6H),8.51(s,6H),8.18(s,6H),7.94(s,6H),7.75(s,6H),7.69(s,6H),7.40–7.38(m,1H),7.40–7.37(m,2H),7.37–7.25(m,28H),6.90(s,6H),6.27(s,3H),5.70(s,6H),4.97(s,3H),4.30(s,6H),3.69(s,5H),3.44(s,5H),1.57(s,18H),1.45(s,6H)。
实施例8
制备N-(2-(3-吡啶基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL05)
除了将苯硼酸换成3-吡啶硼酸之外,其他与实施例4相同方法制备化合物DCL05,产率45%。1H NMR(500MHz,CHCl3)δ9.62(s,4H),8.70(d,J=10.0Hz,8H),8.50(s,4H),8.01(s,4H),7.93(s,4H),7.74(s,3H),7.69(s,5H),7.49(s,1H),7.46(s,5H),7.35(dd,J=59.9,40.0Hz,24H),4.81(s,2H),3.74(s,5H),3.69(s,3H),3.44(s,3H),2.17(s,4H),1.57(s,12H)。
实施例9
制备N-(2-(8-喹啉基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL06)
除了将苯硼酸换成8-喹啉硼酸之外,其他与实施例4相同方法制备化合物DCL06,产率35%。1H NMR(500MHz,CHCl3)δ8.96(d,J=60.0Hz,4H),8.51(s,2H),8.10(s,2H),8.05(s,2H),7.90(s,2H),7.84–7.68(m,6H),7.41–7.28(m,11H),7.30–7.28(m,2H),7.27(s,2H),6.85(s,2H),4.90(s,2H),4.76(s,1H),3.69(s,2H),3.44(s,2H),2.17(s,2H),1.57(s,6H)。
实施例10
制备N-(2-(2-羟基苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL07)
除了将苯硼酸换成2-羟基苯硼酸之外,其他与实施例4相同方法制备化合物DCL07,产率41%。1H NMR(500MHz,CHCl3)δ8.77(s,8H),8.65(s,7H),8.51(s,9H),8.11(s,8H),7.73(d,J=15.0Hz,16H),7.38(d,J=19.5Hz,26H),7.29(dd,J=15.0,10.0Hz,39H),6.97(s,7H),4.94(s,8H),4.52(s,8H),3.57(d,J=125.0Hz,13H),3.41(d,J=3.5Hz,3H),1.57(s,24H),1.48(s,8H)。
实施例11
制备N-(2-(3-羟基苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL08)
除了将苯硼酸换成3-羟基苯硼酸之外,其他与实施例4相同方法制备化合物DCL08,产率41%。1H NMR(500MHz,CHCl3)δ8.51(s,2H),7.99(s,2H),7.93–7.68(m,6H),7.53(s,2H),7.37(d,J=10.8Hz,6H),7.32(s,3H),7.27(s,3H),6.82(s,2H),4.81(s,1H),4.76(s,2H),3.99(s,2H),3.69(s,2H),3.44(s,2H),2.17(s,2H),1.57(s,6H)。
实施例12
制备N-(2-(4-羟基苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL09)
除了将苯硼酸换成4-羟基苯硼酸之外,其他与实施例4相同方法制备化合物DCL09,产率41%。1H NMR(500MHz,CHCl3)δ8.49(s,3H),8.17(s,3H),7.77–7.66(m,12H),7.40(s,3H),7.32(d,J=20.0Hz,11H),7.25(s,2H),6.84(s,6H),4.87(s,3H),4.77(s,3H),4.53(s,3H),3.56(d,J=124.7Hz,5H),3.40(d,J=3.8Hz,1H),1.57(s,9H),1.48(s,3H)。
实施例13
制备N-(2-(2-乙酰氨基苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL10)
除了将苯硼酸换成2-乙酰氨基苯硼酸之外,其他与实施例4相同方法制备化合物DCL10,产率41%。1H NMR(500MHz,CHCl3)δ9.45(s,3H),8.55(s,3H),8.51(s,3H),8.08(s,3H),7.73(d,J=15.0Hz,6H),7.54(s,2H),7.36(dd,J=21.6,16.6Hz,18H),7.27(s,5H),5.00(s,3H),4.35(s,3H),3.57(d,J=125.0Hz,5H),3.41(d,J=3.5Hz,1H),2.18(s,9H),1.57(s,9H),1.48(s,3H)。
实施例14
制备N-(2-(2-氨基苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL11)
除了将苯硼酸换成2-氨基苯硼酸之外,其他与实施例4相同方法制备化合物DCL11,产率41%。1H NMR(500MHz,CHCl3)δ9.15(s,7H),8.51(s,7H),8.04(s,7H),7.73(d,J=15.0Hz,14H),7.45(s,7H),7.39–7.38(m,1H),7.34(d,J=20.0Hz,26H),7.32(s,8H),7.33–7.20(m,24H),6.89(s,10H),4.65(s,6H),4.23(s,14H),3.98(s,7H),3.69(s,7H),3.44(s,7H),1.57(s,21H),1.32(s,7H)。
实施例15
制备N-(2-(2-甲磺酰氨基苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL12)
除了将苯硼酸换成2-甲磺酰氨基-苯硼酸之外,其他与实施例4相同方法制备化合物DCL12,产率41%。1H NMR(500MHz,CHCl3)δ9.15(s,3H),8.51(s,3H),8.23(s,3H),7.73(d,J=15.0Hz,6H),7.40–7.30(m,14H),7.27(s,4H),7.23(s,2H),6.89(s,4H),6.36(s,3H),4.72(s,3H),3.76(d,J=70.0Hz,6H),3.44(s,4H),3.22(s,8H),1.57(s,9H),1.45(s,3H)。
实施例16
制备N-(2-(2-苯甲酰胺基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL13)
除了将苯硼酸换成2-苯甲酰胺基硼酸之外,其他与实施例4相同方法制备化合物DCL13,产率41%。1H NMR(500MHz,CHCl3)δ8.53(d,J=20.0Hz,53H),8.33(d,J=3.1Hz,11H),8.00(s,21H),7.74(d,J=15.0Hz,59H),7.62(s,15H),7.40(s,2H),7.33(dd,J=38.0,18.0Hz,128H),5.71(s,42H),5.02(s,21H),4.22(s,21H),3.57(d,J=125.0Hz,35H),3.41(d,J=3.8Hz,9H),1.57(d,J=3.1Hz,85H)。
实施例17
制备N-(2-(2-氟苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL14)
除了将苯硼酸换成2-氟苯硼酸之外,其他与实施例4相同方法制备化合物DCL14,产率41%。1H NMR(500MHz,CHCl3)δ8.51(s,3H),8.15(d,J=14.4Hz,6H),7.73(d,J=15.0Hz,6H),7.40–7.34(m,15H),7.32(s,5H),7.27 (s,4H),4.92(s,3H),4.17(s,3H),3.56(d,J=125.0Hz,5H),3.41(d,J=3.8Hz,1H),1.57(s,9H),1.50(s,3H)。
实施例18
制备N-(2-(2-甲氧基-5-氯-苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL15)
除了将苯硼酸换成2-甲氧基-4-氯-苯硼酸之外,其他与实施例4相同方法制备化合物DCL15,产率41%。1H NMR(500MHz,CHCl3)δ8.53(d,J=15.0Hz,2H),8.16(s,1H),7.73(d,J=15.0Hz,2H),7.49(s,1H),7.49(s,1H),7.58–7.25(m,7H),7.06(s,1H),5.04(s,1H),3.79(d,J=0.6Hz,4H),3.57(d,J=125.0Hz,2H),2.64(s,1H),1.57(s,3H)。
实施例19
制备N-(2-(2-羟基-5-氯-苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL16)
除了将苯硼酸换成2-羟基-5-氯-苯硼酸之外,其他与实施例4相同方法制备化合物DCL16,产率41%。1H NMR(500MHz,CHCl3)δ8.74(s,4H),8.44(d,J=70.0Hz,8H),8.02(s,4H),7.85(s,8H),7.73(d,J=15.0Hz,8H),7.38(d,J=21.1Hz,7H),7.36–7.34(m,6H),7.32(s,6H),7.27(s,5H),4.68(s,4H),3.69(d,J=0.5Hz,6H),3.44(s,4H),1.57(s,12H),1.47(s,4H)。
实施例20
制备N-(2-(2-羟基-5-氟-苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL17)
除了将苯硼酸换成2-羟基-5-氟-苯硼酸之外,其他与实施例4相同方法制备化合物DCL17,产率41%。1H NMR(500MHz,CHCl3)δ8.51(s,51H),8.30(s,49H),8.00(s,50H),7.82–7.69(m,151H),7.48(d,J=80.9Hz,3H),7.33(dd,J=38.0,18.0Hz,299H),7.12(s,28H),6.95(s,37H),4.83(s,26H),3.95(s,49H),3.69(s,51H),3.44(s,51H),2.19(s,49H),1.57(s,147H)。
实施例21
制备N-(2-(2-羟基-6-氟-苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL18)
除了将苯硼酸换成2-羟基-6-氟-苯硼酸之外,其他与实施例4相同方法制备化合物DCL18,产率41%。1H NMR(500MHz,CHCl3)δ8.51(s,2H),7.99(s,2H),7.74(d,J=15.0Hz,4H),7.52–7.34(m,8H),7.52–7.24(m,16H),6.94(s,1H),6.74(s,2H),4.74(s,1H),3.69(s,2H),3.44(s,2H),2.66(s,2H),1.57(s,6H),1.44(s,2H)。
实施例22
制备N-(2-(2-羟基-4-氟-苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL19)
除了将苯硼酸换成2-羟基-4-氟-苯硼酸之外,其他与实施例4相同方法制备化合物DCL19,产率41%。1H NMR(500MHz,CHCl3)δ8.88(s,67H),8.51(s,70H),7.99(d,J=17.3Hz,121H),7.73(d,J=15.0Hz,139H),7.40(s,4H),7.40–7.25(m,404H),7.17(s,65H),6.87(s,50H),4.75(s,49H),4.04(s,67H),3.69(s,70H),3.44(s,69H),1.55(d,J=18.6Hz,272H)。
实施例23
制备N-(2-(2-羟基-3-氟-苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL20)
除了将苯硼酸换成2-羟基-3-氟-苯硼酸之外,其他与实施例4相同方法制备化合物DCL20,产率41%。1H NMR(500MHz,CHCl3)δ8.51(s,2H),8.42(s,2H),8.03(s,2H),7.73(d,J=15.0Hz,4H),7.40(s,2H),7.40(s,2H),7.55–7.25(m,12H),7.12(s,1H),7.05(s,1H),5.43(s,2H),4.69(s,2H),3.69(d,J=1.1Hz,3H),3.44(s,2H),1.57(s,6H),1.47(s,2H)。
实施例24
制备N-(2-(2-羟基-3,5-二氟-苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL21)
除了将苯硼酸换成2-羟基-3,5-二氟-苯硼酸之外,其他与实施例4相同方法制备化合物DCL21,产率41%。1H NMR(500MHz,CHCl3)δ8.51(s,1H),8.14(s,1H),7.74(d,J=15.0Hz,2H),7.51(s,1H),7.42(s,1H),7.36(s,2H),7.32(s,1H),7.27(s,1H),6.75(s,1H),5.43(s,1H),4.93(s,1H),4.55(s,1H),3.69(s,1H),3.44(s,1H),1.57(s,3H),1.53(s,1H)。
实施例25
制备N-(2-(2-羟基-4,5-二氟苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL22)
除了将苯硼酸换成2-羟基-4,5-二氟-苯硼酸之外,其他与实施例4相同方法制备化合物DCL22,产率41%。1H NMR(500MHz,CHCl3)δ8.48(d,J=32.0Hz,8H),8.02(s,4H),7.73(d,J=15.0Hz,12H),7.41(s,5H),7.41(s,4H),7.55–7.25(m,24H),7.15(s,2H),4.79(s,2H),3.67(d,J=16.5Hz,8H),3.44(s,4H),1.57(s,12H),1.20(s,4H)。
实施例26
制备N-(2-(2-羟基-5-甲基苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL23)
除了将苯硼酸换成2-羟基-5-甲基苯硼酸之外,其他与实施例4相同方法制备化合物DCL23,产率41%。1H NMR(500MHz,CHCl3)δ8.81(s,4H),8.51(s,4H),8.02(d,J=19.2Hz,8H),7.73(d,J=15.0Hz,8H),7.44(s,4H),7.36(s,8H),7.32(s,6H),7.27(s,3H),7.10(s,5H),6.92(s,4H),4.73(s,2H),4.08(s,4H),3.69(s,4H),3.44(s,4H),2.50(s,12H),1.57(s,12H),1.22(s,4H)。
实施例27
制备N-(2-(2-羟基-5-异丙基苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL24)
除了将苯硼酸换成2-羟基-5-异丙基苯硼酸之外,其他与实施例4相同方法制备化合物DCL24,产率41%。1H NMR(500MHz,CHCl3)δ8.84(s,6H),8.51(s,6H),8.16(s,6H),8.01(s,6H),7.73(d,J=15.0Hz,12H),7.38(d,J=18.4Hz,12H),7.29(d,J=25.0Hz,30H),6.97(s,6H),4.96(s,3H),4.07(s,6H),3.69(s,5H),3.44(s,5H),2.87(s,2H),2.28(s,6H),1.57(s,18H),1.20(s,37H)。
实施例28
制备N-(2-(2,6-二羟基苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL25)
除了将苯硼酸换成2,6-二羟基苯硼酸之外,其他与实施例4相同方法制备化合物DCL25,产率41%。1H NMR(500MHz,CHCl3)δ8.49(s,4H),8.11(s,4H),7.72(d,J=15.0Hz,8H),7.41(s,4H),7.33(d,J=20.0Hz,14H),7.26(s, 3H),7.14(s,2H),6.52(s,8H),5.57(s,8H),4.91(s,2H),4.03(s,4H),3.68(s,3H),3.43(s,3H),1.70(s,4H),1.57(s,12H)。
实施例29
制备N-(2-(2,4-二羟基苯基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL26)
除了将苯硼酸换成2,4-二羟基苯硼酸之外,其他与实施例4相同方法制备化合物DCL26,产率41%。1H NMR(500MHz,CHCl3)δ8.48(d,J=32.3Hz,8H),8.15(s,4H),7.94–7.44(m,12H),7.37(s,4H),7.29(d,J=19.9Hz,14H),7.22(s,3H),6.37(s,4H),6.28(s,4H),4.95(s,2H),4.71(s,4H),4.12(s,4H),3.66(s,4H),3.42(s,3H),1.56(s,12H),1.46(s,4H)。
实施例30
制备N-(2-(2-羟基-3-吡啶基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL27)
除了将苯硼酸换成2-羟基-3-吡啶硼酸之外,其他与实施例4相同方法制备化合物DCL27,产率41%。1H NMR(500MHz,CHCl3)δ8.51(s,3H),8.34(s,1H),8.03(s,2H),7.94(s,2H),7.72(d,J=30.9Hz,4H),7.36(s,4H),7.31(d,J=5.0Hz,5H),7.27(s,2H),6.55(s,1H),4.55(s,2H),3.87–3.32(m,6H),3.44(s,2H),3.44(s,2H),1.57(s,6H),1.38(s,2H)。
实施例31
制备N-(2-(4-羟基-3-吡啶基)-3-(3-甲基-5-苯并吡唑基)5-吡啶基)-2-氨基-2-苯乙醇(DCL28)
除了将苯硼酸换成4-羟基-3-吡啶硼酸之外,其他与实施例4相同方法制备化合物DCL28,产率41%。1H NMR(500MHz,CHCl3)δ9.46(s,4H),8.88(s,4H),8.51(s,4H),8.26(s,4H),8.01(s,4H),7.94(s,4H),7.73(d,J=22.8Hz,8H),7.36(s,8H),7.32(s,6H),7.27(s,5H),6.85(s,4H),4.57(s,2H),3.76(d,J=67.0Hz,8H),3.44(s,4H),1.57(s,12H),1.34(s,4H)。
实施例32
除了将3-甲基-5-溴苯并吡唑换成5-溴苯并吡唑以外,其他与实施例2相同方法制备N-叔丁氧羰基-苯并吡唑5-频那醇硼酸酯,产率84%。1H NMR(500MHz,CHCl3)δ9.00(s,1H),8.31(s,1H),1.63(s,9H),1.14(s,12H)。
实施例33
除了将3-甲基-5-溴苯并吡唑换成3-氟-5-溴苯并吡唑以外,其他与实施例2相同方法制备N-叔丁氧羰基-3-氟苯并吡唑-5-频那醇硼酸酯,产率84%。1H NMR(500MHz,CHCl3)δ8.31(s,1H),1.63(s,9H),1.14(s,12H)。
实施例34
除了将3-甲基-7-氟5-溴苯并吡唑换成3-甲基-7-氟-5-溴苯并吡唑以外,其他与实施例2相同方法制备N-叔丁氧羰基-3-甲基7-氟苯并吡唑-5-频那醇硼酸酯,产率84%。1H NMR(500MHz,CHCl3)δ1.63(s,3H),1.57(s,1H),1.14(s,4H)。
实施例35
除了将3-甲基-6-氟-5-溴苯并吡唑换成3-甲基-6-氟-5-溴苯并吡唑以外,其他与实施例2相同方法制备N-叔丁氧羰基-6-氟苯并吡唑-5-频那醇硼酸酯,产率84%。1H NMR(500MHz,CHCl3)δ8.88(s,1H),1.62(s,9H),1.57(s,3H),1.14(s,12H)。
实施例36
除了将N-叔丁氧羰基-3-叔丁氧酰氨基-5-溴苯并吡唑换成3-氨基-5-溴苯并吡唑以外,其他与实施例2相同方法制备N-叔丁氧羰基3-叔丁氧酰氨基-苯并吡唑-5-频那醇硼酸酯,产率84%。1H NMR(500MHz,CHCl3)δ8.32(s,1H),1.63(s,9H),1.49(s,9H),1.14(s,12H)。
实施例37
除了将N-叔丁氧羰基3-甲基-苯并吡唑-5-频那醇硼酸酯换成N-叔丁氧羰基-苯并吡唑-5-频那醇硼酸酯以外,其他与实施例3相同方法制备制备N-(2-氯-3-(5-苯并吡唑基)-5-吡啶基)-2-氨基苯乙醇,产率48%。1H NMR(500MHz,CHCl3)δ8.50(d,J=11.4Hz,4H),8.10(s,2H),7.93(s,2H),7.73(s,1H),7.68(s,3H),7.32(d,J=20.0Hz,7H),7.25(s,3H),4.62(s,2H),3.68(s,2H),3.44(d,J=9.0Hz,4H),1.37(s,2H)。
实施例38
除了将N-叔丁氧羰基-3-甲基-苯并吡唑-5-频那醇硼酸酯换成N-叔丁氧羰基-3-氟苯并吡唑-5-频那醇硼酸酯以外,其他与实施例3相同方法制备制备N-(2-氯-3-(3-氟-5-苯并吡唑基)-5-吡啶基)-2-氨基苯乙醇,产率48%。1H NMR(500MHz,CHCl3)δ8.47(s,4H),8.04(s,4H),7.91(s,4H),7.69(d,J=20.0Hz,8H),7.30(d,J=19.9Hz,14H),7.24(s,5H),4.71(s,2H),3.67(s,4H),3.42(s,4H),3.15(s,4H),1.36(s,4H)。
实施例39
除了将N-叔丁氧羰基-3-甲基-苯并吡唑-5-频那醇硼酸酯换成N-叔丁氧羰基-3-甲基-7-氟苯并吡唑-5-频那醇硼酸酯以外,其他与实施例3相同方法制备制备N-(2-氯-3-(3-甲基-7-氟5-苯并吡唑基)-5-吡啶基)-2-氨基苯乙醇,产率48%。1H NMR(500MHz,CHCl3)δ7.94(s,2H),7.84(s,2H),7.68(s,2H),7.54–7.24(m,12H),4.83(s,1H),3.93(s,2H),3.69(s,2H),3.44(s,2H),1.87(s,2H),1.57(s,6H)。
实施例40
除了将N-叔丁氧羰基-3-甲基-苯并吡唑-5-频那醇硼酸酯换成N-叔丁氧羰基-3-甲基-6-氟苯并吡唑-5-频那醇硼酸酯以外,其他与实施例3相同方法制备制备N-(2-氯-3-(3-甲基-6-氟-5-苯并吡唑基)-5-吡啶基)-2-氨基苯乙醇,产率48%。1H NMR(500MHz,CHCl3)δ8.03(s,3H),7.91(s,3H),7.64(s,3H),7.50(s,4H),7.31(d,J=19.9Hz,11H),7.24(s,4H),4.91(s,3H),4.21(s,3H),3.55(d,J=124.4Hz,5H),3.40(d,J=3.8Hz,1H),1.56(s,9H),1.46(s,3H)。
实施例41
除了将N-叔丁氧羰基-3-甲基-苯并吡唑-5-频那醇硼酸酯换成N-叔丁氧羰基-3-叔丁氧酰氨基-苯并吡唑-5-频那醇硼酸酯以外,其他与实施例3相同方法制备制备N-(2-氯-3-(3-氨基-5-苯并吡唑基)-5-吡啶基)-2-氨基苯乙醇,产率48%。1H NMR(500MHz,CHCl3)δ8.50(s,10H),7.94(s,10H),7.87(s,10H),7.74(s,6H),7.70(s,15H),7.63(s,20H),7.39(dd,J=8.8,7.2Hz,2H),7.49–7.25(m,50H),4.62(s,9H),3.58(d,J=107.0Hz,22H),3.44(s,9H),1.37(s,10H)。
实施例42
制备N-(2-(2-羟基-5-氯苯基)-3-(5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇(DCL29)
除了将N-(2-氯-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基苯乙醇换成N-(2-氯-3-(5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇以外,其他 与实施例19相同方法化合物DCL29,产率39%。1H NMR(500MHz,CHCl3)δ8.69(s,3H),8.46(d,J=2.5Hz,6H),8.33(s,3H),8.03(s,3H),7.81(s,6H),7.70(d,J=14.9Hz,6H),7.30(dd,J=37.7,17.8Hz,18H),4.73(s,2H),4.11(s,3H),3.67(s,3H),3.42(s,3H),1.52(s,3H)。
实施例43
制备N-(2-(2-羟基5-氯苯基)-3-(3-氟5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇(DCL30)
除了将N-(2-氯-3-(3-甲基5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇换成N-(2-氯-3-(3-氟5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇以外,其他与实施例19相同方法化合物DCL30,产率39%。1H NMR(500MHz,CHCl3)δ8.49(s,1H),8.35(s,1H),7.82(d,J=9.5Hz,3H),7.72(d,J=15.0Hz,2H),7.40(s,1H),7.32(d,J=20.0Hz,4H),7.25(s,1H),4.58(s,1H),4.42(s,1H),3.68(s,1H),3.58(s,1H),3.43(s,1H),1.39(s,1H)。
实施例44
制备N-(2-(2-羟基-5-氯苯基)-3-(3-甲基-7-氟-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇(DCL31)
除了将N-(2-氯-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇换成N-(2-氯-3-(3-甲基-7-氟-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇以外,其他与实施例19相同方法化合物DCL31,产率39%。1H NMR(500MHz,CHCl3)δ8.38(d,J=9.5Hz,12H),7.95(s,6H),7.85(s,12H),7.72(s,6H),7.41(s,6H),7.39–7.38(m,1H),7.34(d,J=20.0Hz,23H),7.26(d,J=12.0Hz,12H),4.96(s,3H),4.09(s,6H),3.69(s,5H),3.44(s,5H),1.88(s,6H),1.57(s,18H)。
实施例45
制备N-(2-(2-羟基-5-氯苯基)-3-(3-甲基-6-氟-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇(DCL32)
除了将N-(2-氯-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇换成N-(2-氯-3-(3-甲基-6-氟-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇以外,其他与实施例19相同方法化合物DCL32,产率39%。1H NMR(500MHz,CHCl3)δ8.37(s,48H),8.04(s,50H),7.85(s,96H),7.72(s,49H),7.45(s, 52H),7.38(s,4H),7.40–7.30(m,229H),7.27(s,38H),4.92(s,48H),4.63(s,48H),4.49(s,48H),3.56(d,J=125.0Hz,80H),3.41(d,J=3.5Hz,20H),1.57(s,144H),1.46(s,48H)。
实施例46
制备N-(2-(2-羟基-5-氯苯基)-3-(3-氨基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇(DCL33)
除了将N-(2-氯-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇换成N-(2-氯-3-(3-氨基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇以外,其他与实施例19相同方法化合物DCL33,产率39%。1H NMR(500MHz,CHCl3)δ8.90(s,1H),8.51(s,1H),8.37(s,1H),8.06(s,1H),7.85(d,J=2.8Hz,4H),7.73(d,J=15.0Hz,2H),7.40(s,1H),7.34(d,J=20.0Hz,3H),7.27(s,1H),4.69(s,1H),3.71(d,J=17.4Hz,2H),3.44(s,1H),1.49(s,1H)。
实施例47
制备N-(2-(7-吲哚基)-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇(DCL34)
除了将苯硼酸换成7-吲哚硼酸之外,其他与实施例4相同方法制备化合物DCL34,产率41%。1H NMR(500MHz,CHCl3)δ8.51(s,2H),8.42(d,J=74.9Hz,8H),8.06(s,5H),7.79(s,5H),7.75–7.68(m,15H),7.39(s,6H),7.36–7.23(m,29H),7.12(s,5H),6.55(s,5H),5.03(s,5H),4.17(s,5H),3.56(d,J=124.8Hz,8H),3.41(d,J=4.2Hz,2H),2.06(s,5H),1.57(s,15H)。
实施例48
制备N-(2-(7-苯并咪唑基)-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇(DCL35)
除了将苯硼酸换成7-苯并咪唑硼酸之外,其他与实施例4相同方法制备化合物DCL35,产率41%。1H NMR(500MHz,CHCl3)δ8.54(d,J=35.0Hz,6H),8.18(s,3H),8.06(s,3H),7.73(d,J=15.0Hz,6H),7.63(s,3H),7.41(d,J=3.5Hz,6H),7.33(d,J=20.0Hz,11H),7.26(s,4H),5.07(s,3H),3.90(s,3H),3.56(d,J=124.9Hz,5H),3.41(d,J=4.6Hz,1H),2.61(s,3H),1.57(s,9H)。
实施例49
制备N-(2-(4-哒嗪基)-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇(DCL36)
除了将苯硼酸换成4-哒嗪硼酸之外,其他与实施例4相同方法制备化合物DCL36,产率41%。1H NMR(500MHz,CHCl3)δ9.82(s,4H),9.31(s,4H),8.50(s,4H),8.04(t,J=48.7Hz,12H),7.93(s,4H),7.93(s,4H),7.74(s,4H),7.66(s,4H),7.38(dd,J=8.7,7.4Hz,1H),7.49–7.24(m,20H),4.64(s,2H),4.07(s,4H),3.69(s,4H),3.44(s,4H),2.20(s,4H),1.57(s,12H)。
实施例50
制备N-(2-(5-嘧啶基)-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇(DCL37)
除了将苯硼酸换成5-嘧啶硼酸之外,其他与实施例4相同方法制备化合物DCL37,产率41%。1H NMR(500MHz,CHCl3)δ9.47(s,3H),9.36(s,6H),8.51(s,3H),8.06(s,3H),7.94(s,3H),7.75(s,3H),7.66(s,3H),7.36(s,6H),7.32(s,4H),7.27(s,4H),4.87(s,3H),3.85(s,3H),3.56(d,J=125.0Hz,5H),3.41(d,J=5.1Hz,1H),2.60(s,3H),1.57(s,9H)。
实施例51
制备N-(2-(8-喹啉基)-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇(DCL38)
除了将苯硼酸换成8-喹啉基硼酸之外,其他与实施例4相同方法制备化合物DCL38,产率41%。1H NMR(500MHz,CHCl3)δ8.96(d,J=60.0Hz,39H),8.51(s,20H),8.24(s,19H),8.10(s,20H),7.90(s,20H),7.84–7.68(m,59H),7.48(s,20H),7.40(s,1H),7.39–7.30(m,85H),7.27(s,15H),4.85(s,19H),3.56(d,J=125.0Hz,32H),3.41(d,J=4.8Hz,8H),3.17(s,19H),2.76(s,19H),1.57(s,57H).
实施例52
制备N-(2-(4-吡唑基)-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇(DCL39)
除了将苯硼酸换成4-吡唑基硼酸之外,其他与实施例4相同方法制备化合物DCL39,产率41%。1H NMR(500MHz,CHCl3)8.46(3H,s),8.13(3H,s),7.89(3H,s),7.83(6H,s),7.70(2H,s),7.65(4H,s),7.29(11H,d,J 19.9),7.23 (4H,s),4.81(3H,s),4.28(3H,s),3.54(5H,d,J 124.3),3.39(1H,d,J 3.8),1.54(12H,d,J 18.5)
实施例53
制备N-(2-(N-甲基-4-吡唑基)-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇(DCL40)
除了将苯硼酸换成N-甲基4-吡唑基硼酸之外,其他与实施例4相同方法制备化合物DCL40,产率41%。1H NMR(500MHz,CHCl3)δ8.51(s,2H),8.19(s,2H),7.95(d,J=10.0Hz,4H),7.75(s,2H),7.69(s,2H),7.34(d,J=20.0Hz,7H),7.26(d,J=9.0Hz,5H),5.06(s,2H),4.44(s,2H),3.94(s,6H),3.56(d,J=125.0Hz,4H),3.41(s,1H),1.58(d,J=6.5Hz,8H).
实施例54
制备N-(2-(2-三氟甲基苯基)-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇。(DCL41)
除了将苯硼酸换成2-三氟甲基苯基硼酸之外,其他与实施例4相同方法制备化合物DCL41,产率41%。1H NMR(500MHz,CHCl3)δ8.51(s,5H),8.10(s,5H),7.95(s,5H),7.73(d,J=15.0Hz,15H),7.37(t,J=8.1Hz,22H),7.32(s,11H),7.27(s,7H),4.97(s,5H),4.06(s,5H),3.56(d,J=125.0Hz,8H),3.41(d,J=4.6Hz,2H),2.65(s,5H),1.57(s,15H).
实施例55
制备N-(2-(2,4-二氟苯基)-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇(DCL42)
除了将苯硼酸换成2,4-二氟苯基硼酸之外,其他与实施例4相同方法制备化合物DCL42,1H NMR(500MHz,CHCl3)δ8.50(s,4H),8.11(s,3H),8.03(s,4H),7.72(d,J=15.0Hz,8H),7.55–7.24(m,24H),7.07(s,3H),6.91(s,4H),4.64(s,4H),3.68(d,J=2.9Hz,6H),3.43(s,5H),1.57(s,12H),1.48(s,4H).
实施例56
制备N-(2-(2-羟甲基苯基)-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇。(DCL43)
除了将苯硼酸换成2-羟甲基苯基硼酸之外,其他与实施例4相同方法制备化合物DCL43,1H NMR(500MHz,CHCl3)δ8.49(s,4H),8.02(d,J=6.3 Hz,8H),7.71(d,J=15.0Hz,8H),7.51(s,4H),7.41(d,J=14.4Hz,9H),7.36–7.28(m,18H),7.25(s,3H),5.41(s,4H),4.79(s,2H),4.60(s,8H),3.78(s,4H),3.68(s,3H),3.43(s,4H),2.14(s,4H),1.57(s,12H).
实施例57
制备N-(2-(2-呋喃基)-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇。(DCL44)
除了将苯硼酸换成2-呋喃基硼酸之外,其他与实施例4相同方法制备化合物DCL44,1H NMR(500MHz,CHCl3)δ8.51(s,20H),8.13(s,20H),8.07(s,19H),7.94(s,20H),7.75(s,19H),7.67(s,20H),7.59(s,19H),7.39(dd,J=8.8,7.3Hz,4H),7.51–7.25(m,98H),6.98(s,10H),4.92(s,19H),4.23(s,19H),3.56(d,J=125.0Hz,32H),3.41(d,J=4.8Hz,8H),2.67(s,19H),1.57(s,58H).
实施例58
制备N-(2-(2-噻吩基)-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇。(DCL45)
除了将苯硼酸换成2-噻吩基硼酸之外,其他与实施例4相同方法制备化合物DCL45,1H NMR(500MHz,CHCl3)δ8.51(s,2H),8.13(s,2H),7.94(s,2H),7.74(d,J=12.3Hz,4H),7.56(d,J=5.0Hz,4H),7.36(s,4H),7.32(s,3H),7.27(s,3H),7.15(s,1H),4.64(s,2H),4.01(s,2H),3.69(s,2H),3.44(s,2H),1.57(s,6H),1.44(s,2H).
实施例59
制备N-(2-(2,5二氟苯基)-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇。(DCL46)
除了将苯硼酸换成2,5二氟苯基硼酸之外,其他与实施例4相同方法制备化合物DCL46,1H NMR(500MHz,CHCl3)δ8.49(s,4H),8.14(s,4H),7.88(s,2H),7.72(d,J=15.0Hz,9H),7.51–7.21(m,32H),4.64(s,4H),3.69(d,J=9.9Hz,8H),3.43(s,4H),1.57(s,12H),1.48(s,4H).
实施例60
制备N-(2-(2-氟-5-三氟甲基苯基)-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇。(DCL47)
除了将苯硼酸换成2氟-5三氟甲基苯基硼酸之外,其他与实施例4相同方法制备化合物DCL47,1H NMR(500MHz,CHCl3)δ8.48(s,2H),8.43(s, 2H),7.99(s,2H),7.71(d,J=15.0Hz,4H),7.59(s,1H),7.41–7.23(m,14H),4.94(s,2H),4.44(s,2H),3.55(d,J=124.7Hz,3H),3.40(d,J=4.0Hz,1H),1.57(d,J=7.2Hz,8H).
实施例61
制备N-(4-氟-2-三氟甲基苯基)-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇。(DCL48)
除了将苯硼酸换成4-氟-2-三氟甲基苯基硼酸之外,其他与实施例4相同方法制备化合物DCL48,1H NMR(500MHz,CHCl3)δ8.51(s,3H),8.07(d,J=7.4Hz,5H),7.73(d,J=15.0Hz,6H),7.51(s,3H),7.44–7.25(m,21H),5.06(s,3H),3.80(s,3H),3.56(d,J=125.0Hz,4H),3.41(d,J=4.6Hz,1H),2.69(s,3H),1.57(s,9H).
实施例62
制备N-(2-氟-4-甲基苯基)-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇。(DCL49)
除了将苯硼酸换成2-氟-4-甲基苯基硼酸之外,其他与实施例4相同方法制备化合物DCL49,1H NMR(500MHz,CHCl3)δ8.48(s,3H),8.12(s,3H),7.98(s,3H),7.71(d,J=14.9Hz,6H),7.35(d,J=20.8Hz,7H),7.33–7.18(m,18H),4.88(s,3H),4.54(s,3H),3.55(d,J=124.6Hz,5H),3.40(d,J=3.8Hz,1H),2.49(s,9H),1.56(s,9H),1.47(s,3H).
实施例63
制备N-(2-氟-4-氯苯基)-3-(3-甲基-5-苯并吡唑基)-5-吡啶基)-2-氨基-2-苯乙醇。(DCL50)
除了将苯硼酸换成2-氟-4-氯苯基硼酸之外,其他与实施例4相同方法制备化合物DCL50,1H NMR(500MHz,CHCl3)δ8.50(s,15H),8.33(s,5H),8.03(s,10H),7.72(d,J=15.0Hz,20H),7.46(s,6H),7.42–7.25(m,67H),7.25(s,2H),4.65(s,9H),3.69(d,J=5.7Hz,18H),3.43(s,10H),1.57(s,29H),1.48(s,10H).
细胞生物学活性的测试
1.实验方法:
1.1酶联免疫吸附测定(ELISA)
(1)酶反应底物Poly(Glu,Tyr)4:1用无钾离子的PBS(10mM磷酸钠缓冲液,150mM NaCl,pH7.2-7.4)稀释成20μg/mL,125μL/孔包被酶标板,置37℃反应12-16小时。弃去孔中液体。洗板,用200μL/孔的T-PBS(含0.1%Tween-20的无钾离子的PBS)洗板三次,每次5分钟。于37℃烘箱中干燥酶标板1-2小时。
(2)每孔加入用反应缓冲液(50mM HEPES pH 7.4,50mM MgCl2,0.5mM MnCl2,0.2mM Na3VO4,1mM DTT)稀释的ATP溶液49μL,每孔中加入1μL待测试化合物,再加入50μL用反应缓冲液稀释的c-Met激酶域重组蛋白启动反应,每次实验需设无ATP对照孔两孔。置37℃摇床(100rpm)反应1小时。弃去孔中液体,T-PBS洗板三次。
(3)加入抗体PY99 100μL/孔(抗体用含BSA 5mg/mL的T-PBS 1:500稀释),37℃摇床反应0.5小时。弃去孔中液体,T-PBS洗板三次。
(4)加入辣根过氧化物酶标记的羊抗鼠二抗100μL/孔(抗体用含BSA 5mg/ml的T-PBS 1:2000稀释),37℃摇床反应0.5小时。弃去孔中液体,T-PBS洗板三次。
(5)加入2mg/ml的OPD显色液100μL/孔(用含有0.03%H2O2的0.1M柠檬酸-柠檬酸钠缓冲液(pH=5.4)稀释),25℃避光反应1-10分钟。
(6)加入2M H2SO450μL/孔中止反应,用可调波长式微孔板酶标仪VERSAmax读数,波长为490nm。
(7)结果分析
1.2磺酰罗丹明B(sulforhodamine B,SRB)蛋白染色法
根据细胞生长速率,将处于对数生长期的肿瘤细胞以90μl/孔接种于96孔培养板,贴壁生长24小时再加测试化合物10μl/孔,每个浓度设三复孔,并设生理盐水溶媒对照及无细胞调零孔。细胞在37℃、5%CO2条件下培养72小时然后倾去培养液,用10%冷TCA固定细胞,4℃放置1小时后用蒸馏水洗涤5次,空气中自然干燥。然后加入由1%醋酸配制的SRB(Sigma)4mg/ml溶液100μl/孔,室温中染色15分钟,去染色液,用1%醋酸洗涤5次, 空气干燥。最后加入100μl/孔的Tris溶液,酶标仪560nm波长下测定OD值。按下列公式计算被测物对癌细胞生长的抑制率:抑制率%=(对照组OD值-给药组OD值)/对照组OD值×100%。半数抑制量IC50值采用Logit法计算。
1.3细胞水平检测活性化合物对酪氨酸激酶及其信号通路的抑制活性
将处于对数生长期的细胞接种于6孔板中。待细胞长至半满后,将培养基换为无血清培养基,饥饿细胞24小时。然后加入不同浓度的化合物作用2-6小时。加入50ng/mL的EGF或VEGF刺激15分钟。收集细胞,用冷的PBS(含1mM钒酸钠)洗一次后加入1×SDS凝胶加样缓冲液(50mMTris-HCl(pH 6.8),100mM DTT,2%SDS,10%甘油,1mM钒酸钠,0.1%溴酚蓝)裂解细胞。细胞裂解物在沸水浴中加热10分钟后,于4℃12000rpm离心10分钟。取上清液进行SDS-PAGE电泳,电泳结束后,用半干电转移系统将蛋白转移至硝酸纤维素膜,将硝酸纤维素膜置于封闭液(5%脱脂奶粉稀释于含1mM钒酸钠的TBS)中室温封闭2小时,然后将膜置于一抗溶液中4℃过夜。用含1mM钒酸钠的TBS洗涤三次,每次15min。将膜置于二抗溶液中室温反应1-2小时;同上洗膜三次后,用ECL试剂发色,压片,显影。
1.4所得化合物活性数据
DCL系列化合物细胞活性数据见下表2:
表2
从表2中可以看出,有30余个化合物在N87肿瘤细胞上的抑制活性低于5μM,其中17个化合物在抑制活性低于1μM,具有较好的活性。其中, 化合物DCL16活性最好。该类有较好的肿瘤应用前景,因而具良好的商业价值。
2.实验结果
2.1 DCL16和DCL17分子水平抑制蛋白酪氨酸激酶的活性
体外ELISA实验结果表明DCL16对KDR(VEGFR2)、EGFR、EGFR/T790M/L858R、Abl、ErbB4、ErbB2、FGFR等酪氨酸激酶具有较强的抑制作用,其IC50值分别为3.1nM、6.5nM、8.5nM、2.3nM、5.1nM、18.0nM;对Flt-1、PDGFRβ、PDGFR-α、RET、EPH-A2、IGF1R和FGFR1酪氨酸激酶活性也有抑制作用(表3)。同时DCL17也对以上靶点均具有较好抑制活性。实验数据表明DCL16和DCL17是一个广谱型的酪氨酸激酶抑制剂。
表3.DCL16在分子水平对蛋白酪氨酸激酶活性的抑制
表4.DCL17在分子水平对蛋白酪氨酸激酶活性的抑制
考虑到EGFR、KDR(VEGFR2)、ErbB2(Her2)和FGFR是临床上实体瘤治疗较为确证的抗肿瘤靶点,我们接下来以这四个个激酶为代表来考察DCL16和DC17对激酶的抑制作用。化合物在1μm浓度下对所测试多数酪氨酸激酶活性有不同的抑制作用。
2.2 DCL16对细胞内受体酪氨酸激酶活性的影响
为了确证DCL16对受体酪氨酸激酶的抑制作用,我们进一步研究了DCL16对细胞中受体酪氨酸激酶自身磷酸化的影响。我们选用EGFR高表达的人皮肤磷癌细胞A431和带有EGFR/T790M/L858R突变的人肺癌细胞NCI-H1975考察DCL16对EGFR受体酪氨酸激酶磷酸化的影响。DCL16对KDR受体酪氨酸激酶磷酸化的影响选用VEGFR2高表达的人原代脐静脉内皮细胞HUVEC。DCL16对ErbB2受体酪氨酸激酶磷酸化的影响选用其高表达细胞人乳腺癌BT474细胞。在检测DCL16对生长因子刺激的受体酪氨酸激酶磷酸化的影响的同时,我们还在细胞中检测了DCL16对受体酪氨酸激酶下游主要信号蛋白Erk和AKT磷酸化水平的影响。
2.2.1 DCL16抑制细胞中EGFR的磷酸化
用无血清培养基饥饿A431细胞24小时后,EGFR受体的磷酸化水平均明显下降。此时加入生长因子EGF刺激细胞,可使EGFR受体的磷酸化水平显著 提高。在加入EGF之前,先用DCL16作用细胞2小时,DCL16可以明显抑制EGF诱导的EGFR的酪氨酸磷酸化(图1)。当DCL16浓度为0.04μM时,EGFR的磷酸化即受到显著的抑制,而阳性药Vandetanib在1μM浓度下才明显抑制EGFR的磷酸化;而当DCL16浓度为5μM时,EGFR的磷酸化几乎完全被抑制,同时伴随着Erk1/2、AKT和S6的磷酸化的下调。
图1表示出了DCL16抑制了细胞水平EGFR激酶及下游信号分子的磷酸化。贴壁过夜的A431细胞用无血清培养基饥饿24小时,然后加入不同浓度DCL16处理细胞2h,收样前10min加入生长因子EGF刺激。收集细胞进行Western Blot检测。
2.2.2 DCL16抑制细胞中VEGFR2的磷酸化
此外,DCL16能够抑制HUVEC细胞中VEGFR2的磷酸化。如图2所示,DCL16在0.2μM的剂量下可显著下调HUVEC细胞中VEGFR2的磷酸化水平,DCL16在5μM的剂量下几乎完全抑制HUVEC细胞中VEGFR2的磷酸化水平,同时伴随着Erk和S6磷酸化的下调,但是却上调了AKT磷酸化的水平,其具体机制尚需进一步研究。而阳性对照药Vandetanib虽然对VEGFR2的磷酸化水平的抑制作用强于DCL16,但是在5μM的剂量下几乎对AKT、Erk和S6的磷酸化没有影响。
图2表示出了DCL16抑制了细胞水平VEGFR2激酶及下游信号分子的磷酸化。贴壁过夜的HUVEC细胞用无血清培养基饥饿24小时,然后加入不同浓度DCL16处理细胞6h,收样前10min加入生长因子VEGF刺激。收集细胞进行蛋白质印迹检测。
2.3 DCL16对不同细胞的增殖抑制作用
我们用SRB法测定了DCL16对肿瘤细胞增殖活性的抑制作用。结果表明化合物DCL16对三种肿瘤细胞均有明显的增殖抑制活性,且优于阳性对照药Vandetanib。
表5.DCL16对肿瘤细胞增殖活性的抑制作用
2.4 DCL16对VEGF刺激的HUVEC细胞增殖的抑制
我们检测了DCL16对VEGF刺激的HUVEC细胞增殖的影响。实验结果显示,DCL16对生长因子刺激的HUVEC细胞增殖的抑制作用较为明显,IC50值为0.328μM,优于阳性药Vandetanib的抑制活性(2.543μM)。
表6.DCL16对VEGF刺激的HUVEC细胞增殖活性的抑制作用
2.4 DCL17对SNU16细胞株的增殖抑制作用
附:上述对照所用的阳性化合物文献来源
ABT869:J Hematol Oncol.2009 Jul 30;2:33
Su11248:Clin Cancer Res.2003 Jan;9(1):327-37.
BIBW2992:Expert Opin Pharmacother.2014 Apr;15(6):889-903
Dasatinib:Drug Des Devel Ther.2015 Feb 9;9:773-9
AEW541:Cancer Cell.2004 Mar;5(3):231-9.
AP24534:Cancer Cell.2009 Nov 6;16(5):401-12.
Vandetanib:Expert Opin Investig Drugs.2014 Sep;23(9):1295-303
Erlotinib:N Engl J Med.2005 Jul 14;353(2):123-32.
GDC0068:Clin Cancer Res.2013 Dec 15;19(24):6976-86.
MK2206:Cancer Cell Int.2015 Feb 4;15(1):13.AZD2171:Cancer Cell.2007 Jan;11(1):83-95.
实施例64
DCL16的药代动力学实验
1.给药方案
分别灌胃和静脉注射给予DCL16,给药体积为10ml/kg,药物通过以下方法配置:加5%DMSO充分震荡使其溶解,再加1%助溶剂EL(聚氧乙烯 蓖麻油),其余体积用水补足,配成目标浓度的溶液。试验前禁食12h,自由饮水。给药后2h统一进食。
口服DCL16(20mg/kg),三只SD大鼠,分别在0、15min、30min、60min、2h、4h、6h、8h和24h眼球取血300μl(肝素化管),离心后取血浆,11000rpm离心5min,分离血浆,于–20℃冰箱中冷冻,冻存后用于血药浓度测定。
尾静脉给药DCL16(10mg/kg),三只SD大鼠,分别在0、15min、30min、60min、2h、4h、6h、8h和24h眼球取血300μl(肝素化管),离心后取血浆,11000rpm离心5min,分离血浆,于–20℃冰箱中冷冻,冻存后用于血药浓度测定。
每个时间点6只大鼠,分组及采血时间点见下表7:
表7
按以上设定时间点经大鼠眼球后静脉丛取血0.3mL,置肝素化试管中,11000rpm离心5min,分离血浆,于–20℃冰箱中冷冻。
2.药动学结果
大鼠静脉注射和灌胃给予DCL16后,血浆中药物浓度见表8和表9,相应的药动学参数见表10和表11。
静脉注射给予10mg/kg DCL16后,其在大鼠体内清除率CL为0.48±0.14L/h/kg,AUC0-t为22315±7687ng·h/ml,t1/2为1.24±0.93h。
灌胃给予20mg/kgDCL16后,其在大鼠体内吸收平稳,血浆浓度达峰时间Tmax为1h,达峰浓度Cmax为2228±65ng/ml,血浆浓度-时间曲线下面积AUC0-t为22041±2705ng·h/ml,消除半衰期t1/2为4.52±1.82h。
剂量标准化后,大鼠灌胃给予20mg/kg DCL16后绝对生物利用度为49.4%。
表8 大鼠静脉注射10mg/kg DCL16后的血浆浓度(ng/mL)
表9 大鼠静脉灌胃给予20mg/kg DCL16后的血浆浓度(ng/mL)
表10 大鼠静脉注射10mg/kg DCL16后的药动学参数
表11 大鼠灌胃给予20mg/kg DCL16后的药动学参数
从上面的药代动力学实验中我们发现DCL16灌胃给药条件下具有较好的生物利用度(49.4%),其半衰期也长达4.52小时,且血药浓度较高,有利于减少服药次数与延长药物在体内作用时间。
实施例65
DCL17的药代动力学实验
一、给药方案
健康大鼠6只,体重150-200g,随机分成2组,每组3只。分别灌胃和静脉注射给予DCL17,给药体积为10mL/kg,药物以DMSO/吐温80/生理盐水(5:5:90,v/v/v)配制。试验前禁食12h,自由饮水。给药后2h统一进食。
每个时间点3只小鼠,分组及采血时间点见下表12:
表12
按以上设定时间点经大鼠眼球后静脉丛取血0.3mL,置肝素化试管中,11000rpm离心5min,分离血浆,于–20℃冰箱中冷冻。
二、药动学结果
大鼠灌胃和静脉注射给予DCL17后,血浆中的药物浓度见表13和表14,药动学参数见表15。
大鼠灌胃给予20mg/kg DCL17后,血浆浓度达峰时间Tmax为1h,达峰浓度Cmax为2077.4ng/ml;药时曲线下面积AUC0-t为16629.1ng·h/ml;末端消除半衰期t1/2为8.02h。静脉注射给予10mg/kg DCL17后,AUC0-t为13748.4ng·h/ml;经剂量标准化后,大鼠灌胃给予20mg/kg DCL17后的绝对生物利用度为60.48%。
表13 大鼠灌胃给予20mg/kg DCL17后的血浆浓度(ng/mL)
表14 大鼠静脉注射10mg/kg DCL17后的血药浓度(ng/mL)
表15 大鼠灌胃和静脉注射给予DCL17后的药动学参数
从上面的药代动力学实验中我们发现DCL17灌胃给药条件下具有较好的生物利用度(60.48%),其半衰期也长达5.21小时,且血药浓度较高,有利于减少服药次数与延长药物在体内作用时间。
- 还没有人留言评论。精彩留言会获得点赞!