含有二苯甲基萘的氢化喹啉亚胺镍配合物催化剂及其制备方法与应用与流程
本发明涉及一类含有二苯甲基萘的氢化喹啉亚胺镍配合物催化剂及其制备方法与应用,属于聚乙烯蜡催化剂的设计、合成及应用领域。
背景技术:
聚烯烃主要包括聚乙烯、聚丙烯和聚1-丁烯等,其中聚乙烯树脂(PE)是通用合成树脂中产量最大的品种,并且由于其优良的材料性能,被广泛的应用于生产和生活的各个方面。聚乙烯蜡(又称高分子蜡,简称PE蜡)是低分子量聚乙烯均聚物或共聚体,因其优良的耐寒性、耐热性、耐化学性和耐磨性而得到广泛的应用。正常生产中,这部分蜡作为一种添加剂可直接加到聚烯烃加工中,它可以增加产品的光译和加工性能。作为润滑剂,其化学性质稳定、电性能良好。聚乙烯蜡与高分子量聚乙烯、聚丙烯、聚蜡酸乙烯、乙丙橡胶、丁基橡胶相溶性好。能改善聚乙烯、聚丙烯、ABS的流动性和聚甲基丙烯酸甲酯、聚碳酸酯的脱模性。与PVC的其它外部润滑剂相比,聚乙烯蜡具有更强的内部润滑作用。聚乙烯蜡在溶剂型涂膜中的主要作用为:消光、抗划伤、抗耐磨、抗抛光、抗刻印、防粘连、防沉淀、触变性;良好的润滑性和加工性;金属颜料定位性。聚乙烯蜡的作用原理是:聚乙烯蜡在高温(约100~140℃)下溶解于溶剂中,而在冷却至常温时析出,以微晶形式存在于涂料中,因其触变性有利于涂料的贮存,而在涂料施工应用之后,在溶剂挥发过程中能迁移到涂膜表层,最终与涂料其他组分形成一个“蜡化”的表层。聚乙烯蜡是一类高附加值的聚乙烯材料。
目前,国内通常采用利用裂解的方法制备聚乙烯蜡。该方法成本高、耗能大,而国内炼油厂催化裂化加工装置的生产能力为3.6×107t/a,干气就达数十万吨,其中含有20%~30%乙烯及少量丙烯作为燃料烧掉,造成极大的浪费。所以,直接利用炼油厂干气中的烯烃为原料,将其聚合成低分子聚乙烯(即聚乙烯蜡)是炼油厂提高企业经济效益的一项重要举措,具有广阔的应用前景。美、日、德三国此项技术处于领先地位,日本三井油化公司采用 Ziegler-Natta(齐格勒-纳塔)型催化剂,制得了低分子聚乙烯,其所用主催化剂为钛系催化剂,德国巴斯大公司及美国联碳公司亦开发了同类催化剂,但归纳起来主要是以过渡金属中的铬、钒、锆、钛四大金属化合物为主的催化剂,辅以主催化剂及聚合反应相适应的助催化剂及载体组成催化体系,而关于后过渡金属催化剂制备聚乙烯蜡鲜有报道。烯烃催化剂是决定高分子性能,包括分子量及分子量分布的重要因素,因此开发具有性能改善的聚烯烃催化剂和聚合方法是非常重要的课题。
目前,工业化的聚乙烯催化剂有Ziegler-Natta型催化剂(DE Pat 889229(1953);IT Pat 536899(1955),Phillips型催化剂(Belg.Pat.530617(1955)和茂金属型催化剂(W.Kaminsky,Metalorganic Catalysts for Synthesis and Polymerization,Berlin:Springer,1999),以及近年来发展的后过渡金属配合物型的高效乙烯齐聚和聚合催化剂。
后过渡金属因为具有高的催化活性,主催化剂容易制备和可以通过调节配体及聚合条件来调控聚合物的性质等优势,近些年得到和学术界和工业界的广泛关注。镍配合物催化乙烯齐聚(SHOP工艺,1)是上世纪八十年代后过渡金属催化乙烯反应中具有划时代意义的贡献,基于α-烯烃的大规模生产,极大地推进了化工业的发展(Angew.Chem.,Int.Ed.Engl.,1978,17,466;Angew.Chem.,Int.Ed.Engl.,1983,22,503)。
1995年,Brookhart研究组首次报道了一类新型的α-二亚胺Ni(II)和Pd(II)配合物催化剂2,用于乙烯、α-烯烃和极性单体的聚合与共聚,得到具有独特结构的高分子量聚合物((a)J.Am.Chem.Soc.1995,117,6414;(b)J.Am.Chem.Soc.1996,118,11664)。这一类催化体系是发现的第一类催化乙烯和高级α-烯烃聚合的后过渡金属催化体系,此类催化剂的发现引起了人们对后过渡金属配合物烯烃催化剂研究的兴趣。
由于α-二亚胺配体由于合成简单制备简便,并很容易通过改变与亚胺碳原子相连的取代基调节配体的立体效应和电子效应来改变催化性能,因此一直受到人们的广泛关注。Guan等次报道了基于α-二亚胺环芳亚胺镍催化剂3A具有高的热稳定性,对乙烯聚合表现出高活性,高达4.2×107g(PE)mol-1(Ni)h-1(Angew.Chem.Int.Ed.2004,43,1821)。3B通过在配位中心周围引入配位点,增大空间位阻,降低了β-H消除的速率,减少链转移,降低支化度,催化乙烯聚合可以得到高分子量的高度线性聚乙烯(J.Am.Chem.Soc.,2008,130,7538)。发明人也设计和开发了多种不对称的二苯甲基取代的镍配合物乙烯聚合催化剂(4A,4B):通过调控去取代基的电子效应以及骨架,得到高活性催化剂体系,获得高分子量聚乙烯或双峰分布聚乙烯。
(Organometallics,2011,30,2418;J.Organomet.Chem.,2013,725,37;Catal.Sci.Technol.,2013,3,1172;Appl.Catal.A-Gen.,2014,475,195)。
将α-二亚胺的一个亚胺基换成吡啶,构成的具有相似配位环境的吡啶亚胺类配体也引起了人们的极大兴趣,1999年,Laine首次合成了吡啶单亚胺配体以及相应卤素桥联的镍的配合物5A,发现其对乙烯聚合表现出高活性(6.8×106g(PE)mol-1(Ni)h-1)得到分子量分布窄的聚乙烯(Macromol.Rapid Commun.1999,20,487;Eur.J.Inorg.Chem.1999,959).我们组通过引入在亚胺邻位引入二苯甲基5B,该类配合物催化剂能很好的聚合乙烯,最高活性可达得到1.39×107g(PE)mol-1(Ni)h-1。得到中等支化的低分子量聚 乙烯蜡(Dalton Trans.,2012,41,11999)。当引入二苯甲基萘胺衍生物,得到的配合物5C催化剂均能高效催化乙烯聚合,得到高支化低分子量的聚乙烯蜡(Dalton Trans.,2014,43,423;Dalton Trans.,2014,43,3339)。说明取代基对聚合物的性能影响很大。
除了吡啶亚胺,α-二亚胺体系,我们还设计合成了系列氢化喹啉亚胺镍配合物。6A对乙烯表现出高的聚合活性(Dalton Trans.,2011,40,8436;ACS Catal.,2011,1,1213);6B则表现出高的齐聚活性,齐聚物主要为二聚(C4)和三聚(C6)(New J.Chem.,2011,35,178)。6C表现出高的乙烯聚合活性,产生高支化窄分布的聚乙烯(Dalton Trans.,2012,41,1617)。
纵观近十年来后过渡金属烯烃聚合催化剂的研究结果,后过渡金属配合物催化剂由于具有比茂金属催化剂合成简单、成本低和稳定性好,而且催化剂结构也易于修饰来调控产物(聚合物和齐聚物)结构和分子量的优势越来越受到工业界和学术界的重视。然而作为新的催化剂体系,仍然还有一些基础研究的难点和推进工业化的制约因素。可供选择的催化剂体系(准确地讲具有好活性的催化剂种类)仍然不多,制约了基础研究的发展。其次,由于β-氢链转移终止速率随着温度的升高而增加,以及配合物本身的热稳定性能较差,这势必会引起活性随反应温度升高而降低。如何获得更高活性的乙烯齐聚和聚合催化剂成为研究的核心内容,获得高附加值的聚乙烯材料亦是能 否尽快推进工业化的关键。
本发明人设计合成了一类含有二苯甲基萘的氢化喹啉亚胺镍配合物催化剂,在较高温度下依然能保持很高的乙烯聚合活性,得到窄分布且分子量可调的聚乙烯蜡,具有极大的工业应用价值。
技术实现要素:
本发明的提供一类含有二苯甲基萘的氢化喹啉亚胺镍配合物催化剂及其制备方法与应用。
在本发明的一个方面,提供了一种式IV所示的配体化合物,
其中,R1、R2、R3、R4、R5、R6、R7、R8、R9彼此独立地选自H、F、Cl、Br、I或任选被一个或多个R’取代的下列基团:C1-6烷基-、C1-6烷基氧基-、C3-10环烷基-、C3-10环烷基氧基-、C6-14芳基-、C6-14芳基氧基-;
每个R’独立地选自F、Cl、Br、I、C1-6烷基-、C1-6烷基氧基-、C3-10环烷基-、C3-10环烷基氧基-、C6-14芳基-、C6-14芳基氧基-;
i为1、2、3、4、5或6,j和k彼此独立地为1、2、3、4或5。
在本发明的示例性实施方案中,R1和R4彼此独立地选自H或任选被一个或多个R”取代的下列基团:C6-14芳基C1-6烷基-、二C6-14芳基C1-6烷基-;
优选地,R1和R4彼此独立地选自H或任选被一个或多个R”取代的下列基团:苯基甲基、苯基乙基、二苯基甲基、二苯基乙基、萘基甲基、萘基乙基、二萘基甲基、二萘基乙基;
每个R”独立地选自F、Cl、Br、I、C1-6烷基-、C1-6烷基氧基-、C6-14芳基-、C6-14芳基氧基-,优选F、Cl、Br、I、甲基、乙基、丙基、异丙基、甲 氧基、乙氧基、苯基、苯氧基。
作为实例,式IV的化合物选自具有如下基团定义的化合物:
L1:R1~R9=H;
L2:R1=Ph2CH;R2~R9=H;
L3:R1=R4=Ph2CH;R2=R3=R5=R6=R7=R8=R9=H;
L4:R4=Ph2CH;R1=R2=R3=R5=R6=R7=R8=R9=H。
本发明提供的式I所示的含有二苯甲基萘的氢化喹啉亚胺镍配合物,
其中,R1、R2、R3、R4、R5、R6、R7、R8、R9、i、j和k彼此独立地具有上文所述的定义;
X为氯或溴。
作为示例,式I配合物选自具有如下基团定义的配合物:
Ni1:R1~R9=H;X=Br;
Ni2:R1=Ph2CH;R2~R9=H;X=Br;
Ni3:R1=R4=Ph2CH;R2=R3=R5=R6=R7=R8=R9=H;X=Br;
Ni4:R4=Ph2CH;R1=R2=R3=R5=R6=R7=R8=R9=H;X=Br;
Ni5:R1~R9=H;X=Cl;
Ni6:R1=Ph2CH;R2~R9=H;X=Cl;
Ni7:R1=R4=Ph2CH;R2=R3=R5=R6=R7=R8=R9=H;X=Cl;
Ni8:R4=Ph2CH;R1=R2=R3=R5=R6=R7=R8=R9=H;X=Cl。
本发明还提供了一种用于乙烯聚合的催化剂组合物,包括作为活性组分的式I所示的配合物,以及任选的助催化剂。
所述助催化剂可以选自例如甲基铝氧烷、改性的甲基铝氧烷(MMAO)和氯化二乙基铝(DEAC)中的一种或多种。
根据本发明,所述改性的甲基铝氧烷为三甲基铝与甲基铝氧烷的混合物。所述改性的甲基铝氧烷可通过例如水解三甲基铝制备甲基铝氧烷来制备:三甲基铝水解后,残留部分未水解的三甲基铝与甲基铝氧烷的混合物称为改性的。
优选地,助催化剂为改性的甲基铝氧烷或氯化二乙基铝;所述催化剂组合物中,铝氧烷中金属Al与主催化剂中心金属Ni的摩尔比为1000~1750:1,优选摩尔比为1250:1;氯化二乙基铝中金属Al与主催化剂中心金属Ni的摩尔比为300~450:1,优选摩尔比为400:1。
本发明还提供了式IV化合物的制备方法,包括在催化剂的存在下,将式II所示的5,6,7-三氢喹啉-8-酮类化合物与式III所示的二苯甲基取代的萘胺衍生物进行缩合反应,得到所述式IV所示的配体化合物:
其中,式II和III中R1~R9的定义彼此独立地具有上述式IV中的定义。
对于缩合反应的具体条件和参数没有特别限制,本领域技术人员可以根据具体情况选择具体的反应条件和参数,以制备目标化合物。以下示例性地描述了该反应可用的条件和参数,本领域技术人员可以将其中一个或多个条件和参数应用于该反应,或者,也可以对它们进行进一步的变型或调整。
所述缩合反应可以使用本领域中任何适宜的催化剂,其中的一个实例为对甲苯磺酸;
对于催化剂的用量没有特别限定,只要其达到催化效果即可。作为示例,所述催化剂的用量为催化量,例如反应底物式II或III化合物摩尔量的5%-10%;
式II化合物与式III化合物的投料摩尔比可以为1.0~1.5:1.0~1.5,例如为1.0:1.0~1.2或1.0~1.2:1.0,具体可以为1.0:1.1或1.1:1.0;
反应可以在有机溶剂,例如苯或甲苯中进行;
反应温度可以在较宽的范围内选择。优选地,可以在共沸温度或回流温度反应以除去反应中生成的水。作为示例,当溶剂为甲苯时,反应温度为120℃;
反应时间可以为例如4-8小时。
上述缩合反应完毕后,所得式IV所示的配体化合物可以进一步纯化。所述纯化方法可以包括如下步骤:
1)将所得式IV所示的配体化合物溶于有机溶剂(如二氯甲烷)中;
2)使用碱性氧化铝担载进行柱层析,以石油醚和乙酸乙酯的混合溶剂为淋洗剂进行洗脱,通过薄层色谱检测洗脱流分;
3)除去溶剂,得到纯化的式IV所示配体化合物。
本发明还提供式I配合物的制备方法,包括如下步骤:
根据本发明,上述反应可以在适宜的有机溶剂中进行。所述有机溶剂可以选自例如甲醇、乙醇、一氯甲烷、二氯甲烷、三氯甲烷中的一种或多种, 优选乙醇、二氯甲烷或其混合物。
式IV所示配体化合物与(DME)NiBr2或NiCl2·6H2O的摩尔比可以为0.9~1.1:0.9~1.1,具体为1:1;
反应温度可以为室温,时间可以为12-24小时;
作为示例,所述配合物的制备方法包括在室温下,将式IV所示配体化合物加入到二氯甲烷和乙醇的混合溶剂中,加入(DME)NiBr2或NiCl2·6H2O混匀进行络合反应,得到式I的配合物。
本发明还提供了一种制备聚乙烯的方法,包括在前述催化剂组合物的存在下使乙烯进行聚合反应。
优选地,所述聚合反应步骤中,温度可以为20-60℃;聚合压力可以为10atm;时间可以为10min-60min;
所述聚合反应可以在溶剂中,优选甲苯进行;
所述聚合反应可以在惰性气氛,优选氮气、氩气或其混合物中进行。
本发明还提供上述式I配合物或上述催化剂组合物在催化烯烃聚合,特别是乙烯聚合反应中的应用。
术语定义和解释
术语“C1-6烷基”应理解为优选表示具有1、2、3、4、5或6个碳原子的线性的或支化的饱和一价烃基,例如甲基、乙基、丙基、丁基、戊基、己基、异丙基、异丁基、仲丁基、叔丁基、异戊基、2-甲基丁基、1-甲基丁基、1-乙基丙基、1,2-二甲基丙基、新戊基、1,1-二甲基丙基、4-甲基戊基、3-甲基戊基、2-甲基戊基、1-甲基戊基、2-乙基丁基、1-乙基丁基、3,3-二甲基丁基、2,2-二甲基丁基、1,1-二甲基丁基、2,3-二甲基丁基、1,3-二甲基丁基或1,2-二甲基丁基或它们的异构体。特别地,所述基团具有1、2、3或4个碳原子(“C1-4烷基”),例如甲基、乙基、丙基、丁基、异丙基、异丁基、仲丁基、叔丁基,更特别地,所述基团具有1、2或3个碳原子(“C1-3烷基”),例如甲基、乙基、正丙基或异丙基。
术语“C3-10环烷基”应理解为表示饱和的一价单环或双环烃环,其具有3、4、5、6、7、8、9或10个碳原子。所述C3-10环烷基可以是单环烃基, 如环丙基、环丁基、环戊基、环己基、环庚基、环辛基、环壬基或环癸基,或者是双环烃基如十氢化萘环。
术语“C6-14芳基”应理解为优选表示具有6、7、8、9、10、11、12、13或14个碳原子的一价芳香性或部分芳香性的单环、双环或三环烃环(“C6-14芳基”),特别是具有6个碳原子的环(“C6芳基”),例如苯基;或联苯基,或者是具有9个碳原子的环(“C9芳基”),例如茚满基或茚基,或者是具有10个碳原子的环(“C10芳基”),例如四氢化萘基、二氢萘基或萘基,或者是具有13个碳原子的环(“C13芳基”),例如芴基,或者是具有14个碳原子的环(“C14芳基”),例如蒽基。
本申请说明书和权利要求书描述的结构式中,取代基R7、R8、R9连接的虚线“--------”表示可以在相应环系的任何取代位置上取代。具体而言,所述取代基可以在苯环的邻、间、对位,也可以在氢化喹啉的2-、3-、4-、5-、6-、7-位上取代。
本发明设计并合成了含有N^N两齿配位卤素(Br或Cl)桥联的二苯甲基萘的氢化喹啉亚胺镍配合物,该类金属配合物用于催化乙烯聚合反应,表现出高的催化活性,可达到9.39×106g·mol-1(Ni)·h-1,同时得到高度支化的聚乙烯,并且该类配合物催化剂在较高温度下(50℃)仍可以保持较高的持久的活性,具有广泛的工业应用前景。
附图说明
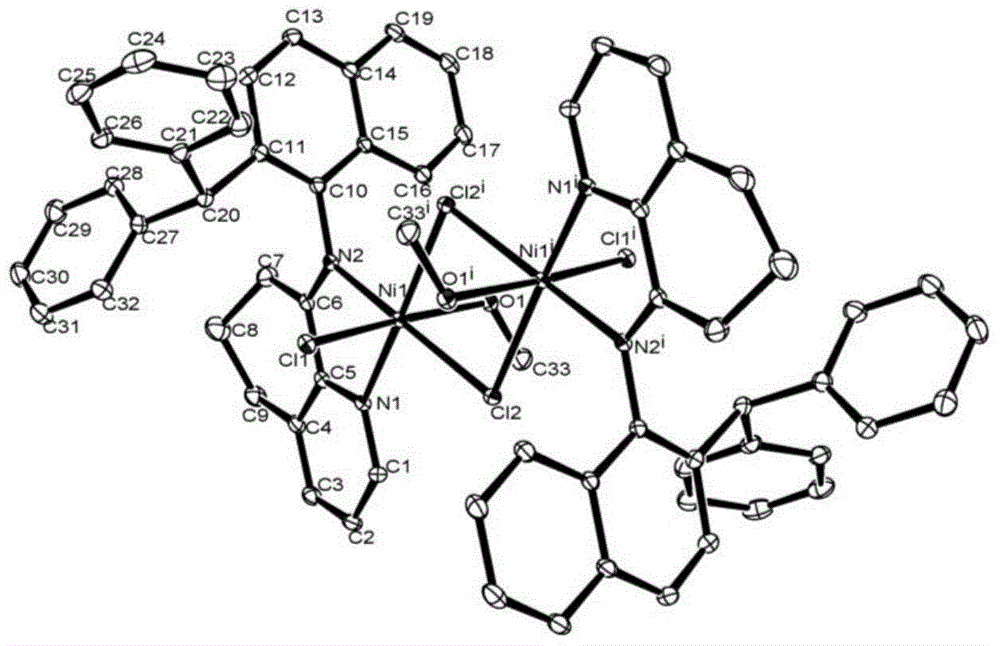
图1为配合物Ni1的晶体结构示意图;
图2为配合物Ni5的晶体结构示意图。
具体实施方式
下面结合具体实施例对本发明作进一步阐述,但本发明并不限于以下实施例。
所述方法如无特别说明均为常规方法。所述原材料如无特别说明均能从公开商业途径而得。
下述乙烯聚合实施例中所得聚合物的分子量及分子量分布均为按照常规的GPC方法测定而得,熔点均为按照常规的DSC方法测定而得,聚合物 的聚合活性均按照如下公式计算而得:聚合活性=聚合物产量/(催化剂用量·聚合时间)。支化度的计算方法参考文献(Macromolecules 1999,32,1620–1625;Polym.J.1984,16,731–738).
实施例1、制备8-(2-二苯甲基萘亚胺)-5,6,7-三氢喹啉的合成方法(L1)
1)将5,6,7-三氢喹啉-8-酮(0.882g,5.99mmol)和2-二苯甲基-1-萘胺(2.04g,6.59mmol)溶于120mL甲苯,催化量对甲苯磺酸(0.228g,1.20mmol)加入反应液,反应液加热回流8h。碱性氧化铝担载柱层析分离得到0.97g黄色粉末,产率为37%(熔点:165–166℃)。
结构确证数据如下:
1H NMR(400MHz,CDCl3,TMS):δH 8.80(d,1H,J=3.2Hz,Py–H),7.79(d,1H,J=8.0Hz,Py–H),7.57(d,1H,J=8.4Hz,Py–H),7.50(d,2H,J=8.4Hz,Ar–H),7.41–7.08(m,14H,Ar–H),5.92(s,1H,–CHPh2),2.72–2.64(m,1H,–CH2),2.53–2.46(m,1H,–CH2),1.90–1.83(m,1H,–CH2),1.20–1.13(m,1H,–CH2),0.88–0.77(m,2H,–CH2).
13C NMR(100MHz,CDCl3,TMS):167.8,149.8,148.9,146.2,144.5,142.9,137.5,137.2,132.8,130.2,129.7,128.6,128.3,128.1,127.6,126.1,125.5,125.0,123.5,122.5,51.8,31.4,29.2,21.2.
FT–IR(KBr,disk,cm-1):3054,3026,1641(νC=N),1564,1488,1450,1428,1375,1315,1199,1097,1028,790,745,701.
元素分析:C32H26N2(438.21)理论值:C,87.64;H,5.98;N,6.39%;实测值:C,87.48;H,6.22;N,6.27%.
实施例2、制备8-[2,4-二(二苯甲基)萘亚胺]-5,6,7-三氢喹啉的合成方法(L2)
使用与实施例1相同方法得到0.81g黄色固体,产率为36%(熔点:118–119℃)。
结构确证数据如下:
1H NMR(400MHz,CDCl3,TMS):δH 8.80(d,1H,J=3.2Hz,Py–H),7.94(d,1H,J=8.0Hz,Py–H),7.60(d,1H,J=8.0Hz,Py–H),7.52(d,1H,J=7.2Hz,Ar–H),7.34–6.88(m,23H,Ar–H),6.63(s,1H,Ar–H),6.16(s,1H,–CHPh2),5.80(s,1H,–CHPh2),2.74–2.67(m,1H,–CH2),2.56–2.49(m,1H,–CH2),1.93–1.86(m,1H,–CH2),1.27–1.20(m,2H,–CH2),0.98–0.97(m,1H, –CH2).
13C NMR(100MHz,CDCl3,TMS):167.9,149.8,148.9,145.0,144.3,137.5,137.3,134.0,130.3,130.1,129.7,129.4,129.3,128.4,128.3,127.9,126.2,126.0,125.8,125.7,125.1,125.0,124.6,124.5,53.0,51.8,31.6,29.3,21.3.
FT–IR(KBr,disk,cm-1):3053,3025,1643(νC=N),1577,1491,1450,1381,1327,1190,1108,1075,1031,916,796,743,697.元素分析:C45H36N2(604.29)
理论值:C,89.37;H,6.00;N,4.63%;实测值:C,89.22;H,6.24;N,4.52%.
实施例3、制备8-[2,4,7-三(二苯甲基)萘亚胺]-5,6,7-三氢喹啉的合成方法(L3)
使用与实施例1相同方法得到1.34g黄色固体,产率为38%(熔点:185–186℃)。
结构确证数据如下:
1H NMR(400MHz,CDCl3,TMS):δH 8.74(d,1H,J=3.2Hz,Py–H),7.87(d,1H,J=8.8Hz,Py–H),7.48(d,1H,J=7.2Hz,Py–H),7.31–6.89(m,33H,Ar–H),6.59(s,1H,Ar–H),6.13(s,1H,–CHPh2),5.77(s,1H,–CHPh2),5.53(s,1H,–CHPh2),2.51–2.47(m,2H,–CH2),1.47–1.42(m,1H,–CH2),1.10–1.07(m,2H,–CH2),0.72–0.69(m,1H,–CH2).
13C NMR(100MHz,CDCl3,TMS):167.8,148.6,144.2,144.1,143.8,143.6,143.2,140.3,137.2,137.0,133.8,130.0,129.6,129.4,129.3,128.3,128.2,128.0,127.9,127.7,126.2,126.0,125.9,125.8,124.8,124.6,124.4,124.1,56.8,53.0,51.9,31.5,29.2,21.1.
FT–IR(KBr,disk,cm-1):3051,3023,1640(νC=N),1598,1563,1489,1437,1368,1375,1319,1250,1103,1075,1028,910,789,745,695.
元素分析:C58H46N2(770.37)理论值:C,90.35;H,6.01;N,3.63%;实测值:C,90.15.;H,6.27;N,3.56%.
实施例4、制备8-(2-二苯甲基萘亚胺)-5,6,7-三氢喹啉溴化镍的合成方法(Ni1)
将实施例1所得8-(2-二苯甲基萘亚胺)-5,6,7-三氢喹啉(L1)(0.219g,0.5mmol)与(DME)NiBr2(0.154g,0.5mmol)在CH2Cl2(5mL)和EtOH(5mL)的混合溶液中室温搅拌24h.反应液浓缩,加入大量乙醚沉淀,沉淀物 通过过滤收集,并用大量乙醚洗涤。得到黄色粉末0.28g,产率为85%。
FT–IR(KBr,disk,cm-1):3055,2969,1617(νC=N),1583,1493,1451,1333,1217,1192,1131,809,750,727,702.
元素分析:C32H26Br2N2Ni(653.98)理论值:C,58.49;H,3.99;N,4.26%.实测值:C,58.27;H,4.12;N,4.02%.
该化合物的晶体结构图如图1所示;由图1可知,金属镍中心与二个氮原子和一个甲醇配位通过两个溴原子桥联形成双核六配位的配合物;中心原子采取了扭曲的八面体的几何构型。
实施例5、制备8-[2,4-二(二苯甲基)萘亚胺]-5,6,7-三氢喹啉溴化镍的合成方法(Ni2)
将实施例2所得8-[2,4-二(二苯甲基)萘亚胺]-5,6,7-三氢喹啉(L2)(0.302g,0.5mmol)与(DME)NiBr2(0.154g,0.5mmol)在CH2Cl2(5mL)和EtOH(5mL)的混合溶液中室温搅拌24h.反应液浓缩,加入大量乙醚沉淀,沉淀物通过过滤收集,并用大量乙醚洗涤。得到黄色粉末0.34g,产率为83%.
FT–IR(KBr,disk,cm-1):3058,3025,1618(νC=N),1588,1493,1450,1336,1219,1124,1075,1030,791,741,698.
元素分析:C45H36Br2N2Ni(820.06)理论值:C,65.65;H,4.41;N,3.40%.实测值:C,65.38;H,4.64;N,3.28%.
实施例6、制备8-[2,4,7-三(二苯甲基)萘亚胺]-5,6,7-三氢喹啉溴化镍的合成方法(Ni3)
将实施例3所得8-[2,4,7-三(二苯甲基)萘亚胺]-5,6,7-三氢喹啉(L3)(0.386g,0.5mmol)与(DME)NiBr2(0.154g,0.5mmol)在CH2Cl2(5mL)和EtOH(5mL)的混合溶液中室温搅拌24h.反应液浓缩,加入大量乙醚沉淀,沉淀物通过过滤收集,并用大量乙醚洗涤。得到黄色粉末0.35g,产率为71%。
FT–IR(KBr,disk,cm-1):3059,3025,1621(νC=N),1592,1494,1451,1354,1336,1125,1076,1031,832,789,745,699.
元素分析:C58H46Br2N2Ni(986.14)理论值:C,70.40;H,4.69;N,2.83%.实测值:C,70.28;H,4.84;N,2.62%.
实施例7、制备8-(2-二苯甲基萘亚胺)-5,6,7-三氢喹啉氯化镍的合成方法(Ni5)
将实施例1所得8-(2-二苯甲基萘亚胺)-5,6,7-三氢喹啉(L1)(0.219g,0.5mmol)与NiCl2·6H2O(0.137g,0.5mmol)在CH2Cl2(5mL)和EtOH(5mL)的混合溶液中室温搅拌24h.反应液浓缩,加入大量乙醚沉淀,沉淀物通过过滤收集,并用大量乙醚洗涤。得到黄色粉末0.25g,产率为88%。
FT–IR(KBr,disk,cm-1):3055,2966,1624(νC=N),1583,1493,1450,1333,1215,1192,1130,809,750,728,703.
元素分析:C32H26Cl2N2Ni(566.08)理论值:C,67.45;H,4.61;N,4.93%.实测值:C,67.35;H,4.82;N,4.75%.
该化合物的晶体结构图如图2所示;由图2可知,金属镍中心与二个氮原子和一个甲醇配位通过两个氯原子桥联形成双核六配位的配合物;中心原子采取了扭曲的八面体的几何构型。
实施例8、制备8-[2,4-二(二苯甲基)萘亚胺]-5,6,7-三氢喹啉氯化镍的合成方法(Ni5)
将实施例2所得8-[2,4-二(二苯甲基)萘亚胺)-5,6,7-三氢喹啉(L2)(0.302g,0.5mmol)与NiCl2·6H2O(0.137g,0.5mmol)在CH2Cl2(5mL)和EtOH(5mL)的混合溶液中室温搅拌24h.反应液浓缩,加入大量乙醚沉淀,沉淀物通过过滤收集,并用大量乙醚洗涤。得到黄色粉末0.32g,产率为88%。
FT–IR(KBr,disk,cm-1):3057,3025,1619(νC=N),1586,1493,1451,1335,1218,1124,1076,1032,791,739,697.
元素分析:C45H36Cl2N2Ni(732.16)理论值:C,73.60;H,4.94;N,3.81%.实测值:C,73.38;H,5.02;N,3.64%.
实施例9、制备8-[2,4,7-三(二苯甲基)萘亚胺)-5,6,7-三氢喹啉氯化镍的合成方法(Ni6)
将实施例3所得8-[2,4,7-三(二苯甲基)萘亚胺]-5,6,7-三氢喹啉(L3)(0.386g,0.5mmol)与NiCl2·6H2O(0.137g,0.5mmol)在CH2Cl2(5mL)和EtOH(5mL)的混合溶液中室温搅拌24h.反应液浓缩,加入大量乙醚沉淀,沉淀物通过过滤收集,并用大量乙醚洗涤。得到黄色粉末0.31g,产率 为70%。
FT–IR(KBr,disk,cm-1):3058,3025,1622(νC=N),1593,1493,1450,1353,1124,1076,1032,791,745,699.
元素分析:C58H46Cl2N2Ni(898.24)理论值:C,77.35;H,5.15;N,3.11%.实测值:C,77.15;H,5.26;N,3.02%.
实施例10、利用配合物Ni2及MMAO联合催化加压下的乙烯聚合:
a)加压下的乙烯聚合使用一台装备有机械搅拌桨和温度控制装置的250毫升不锈钢聚合釜。将聚合釜在烘箱中加热到100℃,加热时间持续两小时。快速取出聚合釜放入保温套管中,安装好反应釜。抽真空用氮气置换3次,同时控温至所需温度。待温度接近所需温度时,在氮气保护下,先用30mL甲苯润洗管道,再将40ml的催化剂Ni2(2μmol)的甲苯溶液和1.1ml的助催化剂MMAO(1.93mol/L,甲苯中)以及剩余用来冲洗管道的30mL甲苯依次加入到250ml不锈钢高压釜中,使得甲苯总体积为100ml。此时Al/Ni=1000:1。机械搅拌开始,保持400转/分,当聚合温度达到30℃时,往反应釜中充入乙烯,聚合反应开始。在30℃下保持10atm的乙烯压强,搅拌30min。用5%盐酸酸化的乙醇溶液中和反应液,得到聚合物沉淀,用乙醇洗数次,真空烘干至恒重,称重得4.72g聚合物,聚合活性:4.72×106g/mol(Ni)h-1,聚合物Tm=103.8℃(Tm为聚合物的熔融温度,通过示差扫描量热法DSC测试所得)。聚合物Mw=4.63kg·mol-1,Mw/Mn=3.88(Mw和Mw/Mn为聚合物的重均分子量和分子量分布,通过凝胶渗透色谱GPC测试所得)。
b)基本同a),区别在于:助催化剂用量为1.3ml的MMAO(1.93mol/L,甲苯中),使Al/Ni=1250:1。聚合活性:7.65×106g/mol(Ni)h-1,聚合物Tm=103.9℃。聚合物Mw=3.42kg·mol-1,Mw/Mn=3.60。
c)基本同a),区别在于:助催化剂用量为1.6ml的MMAO(1.93mol/L,甲苯中),使Al/Ni=1500:1。聚合活性:5.04×106g/mol(Ni)h-1,聚合物Tm=104.2℃。聚合物Mw=3.33kg·mol-1,Mw/Mn=3.11。
d)基本同a),区别在于:助催化剂用量为1.8ml的MMAO(1.93mol/L,甲苯中),使Al/Ni=1750:1。聚合活性:4.57×106g/mol(Ni)h-1,聚合物Tm=105.9℃。聚合物Mw=2.82kg·mol-1,Mw/Mn=2.45。
e)基本同a),区别在于:聚合温度为20℃。助催化剂用量为1.3ml的MMAO(1.93mol/L,甲苯中),使Al/Ni=1250:1。聚合活性:5.14×106g/mol(Ni)h-1,聚合物Tm=106.2℃。聚合物Mw=5.80kg·mol-1,Mw/Mn=4.22。支化度为44/1000C。
f)基本同a),区别在于:聚合温度为40℃。助催化剂用量为1.3ml的MMAO(1.93mol/L,甲苯中),使Al/Ni=1250:1。聚合活性:4.44×106g/mol(Ni)h-1,聚合物Tm=103.8℃。聚合物Mw=3.21kg·mol-1,Mw/Mn=3.33。
g)基本同a),区别在于:聚合温度为50℃。助催化剂用量为1.3ml的MMAO(1.93mol/L,甲苯中),使Al/Ni=1250:1。聚合活性:2.59×106g/mol(Ni)h-1,聚合物Tm=101.1℃。聚合物Mw=2.23kg·mol-1,Mw/Mn=2.97。支化度为79/1000C。
h)基本同a),区别在于:主催化剂为Ni2,聚合温度为30℃。使Al/Ni=1250:1。聚合时间为10min。聚合活性:9.39×106g/mol(Ni)h-1,聚合物Tm=102.5℃。聚合物Mw=2.67kg·mol-1,Mw/Mn=3.35。
i)基本同a),区别在于:主催化剂为Ni2,聚合温度为30℃。使Al/Ni=1250:1。聚合时间为20min。聚合活性:8.30×106g/mol(Ni)h-1,聚合物Tm=103.8℃。聚合物Mw=3.20kg·mol-1,Mw/Mn=3.44。
j)基本同a),区别在于:主催化剂为Ni2,聚合温度为30℃。使Al/Ni=1250:1。聚合时间为30min。聚合活性:7.65×106g/mol(Ni)h-1,聚合物Tm=103.9℃。聚合物Mw=3.42kg·mol-1,Mw/Mn=3.60。
k)基本同a),区别在于:主催化剂为Ni2,聚合温度为30℃。使Al/Ni=1250:1。聚合时间为40min。聚合活性:6.62×106g/mol(Ni)h-1,聚合物Tm=102.7℃。聚合物Mw=3.73kg·mol-1,Mw/Mn=4.27。
l)基本同a),区别在于:主催化剂为Ni2,聚合温度为30℃。使Al/Ni=1250:1。聚合时间为60min。聚合活性:4.77×106g/mol(Ni)h-1,聚合物Tm=103.6℃。聚合物Mw=4.26kg·mol-1,Mw/Mn=4.48。
实施例11、利用配合物Ni1及MMAO联合催化加压下的乙烯聚合
基本同b),区别在于:主催化剂为Ni1;助催化剂用量为1.3ml的MMAO(1.93mol/L,甲苯中),使Al/Ni=1250:1;聚合温度为30℃。聚合活性:7.47 ×106g/mol(Ni)h-1,聚合物Tm=87.1℃。聚合物Mw=1.97kg·mol-1,Mw/Mn=2.19。
实施例12、利用配合物Ni3及MMAO联合催化加压下的乙烯聚合
基本同b),区别在于:主催化剂为Ni3;助催化剂用量为1.3ml的MMAO(1.93mol/L,甲苯中),使Al/Ni=1250:1;聚合温度为30℃。聚合活性:4.68×106g/mol(Ni)h-1,聚合物Tm=71.2℃。聚合物Mw=0.78kg·mol-1,Mw/Mn=1.68。
实施例13、利用配合物Ni5及MMAO联合催化加压下的乙烯聚合
基本同b),区别在于:主催化剂为Ni5;助催化剂用量为1.3ml的MMAO(1.93mol/L,甲苯中),使Al/Ni=1250:1;聚合温度为30℃。聚合活性:6.53×106g/mol(Ni)h-1,聚合物Tm=91.4℃。聚合物Mw=1.96kg·mol-1,Mw/Mn=2.08。
实施例14、利用配合物Ni5及MMAO联合催化加压下的乙烯聚合
基本同b),区别在于:主催化剂为Ni5;助催化剂用量为1.3ml的MMAO(1.93mol/L,甲苯中),使Al/Ni=1250:1;聚合温度为30℃。聚合活性:8.29×106g/mol(Ni)h-1,聚合物Tm=94.9℃。聚合物Mw=2.00kg·mol-1,Mw/Mn=2.10。
实施例15、利用配合物Ni6及MMAO联合催化加压下的乙烯聚合
基本同b),区别在于:主催化剂为Ni6;助催化剂用量为1.3ml的MMAO(1.93mol/L,甲苯中),使Al/Ni=1250:1;聚合温度为30℃。聚合活性:5.50×106g/mol(Ni)h-1,聚合物Tm=64.8℃。聚合物Mw=0.77kg·mol-1,Mw/Mn=2.26。
实施例16、利用配合物Ni2及Et2AlCl联合催化加压下的乙烯聚合
a)加压下的乙烯聚合使用一台装备有机械搅拌桨和温度控制装置的250毫升不锈钢聚合釜。将聚合釜在烘箱中加热到100℃,加热时间持续两小时。快速取出聚合釜放入保温套管中,安装好反应釜。抽真空用氮气置换3次, 同时控温至所需温度。待温度接近所需温度时,在氮气保护下,先用30mL甲苯润洗管道,再将40ml的催化剂Ni2(2μmol)的甲苯溶液和1.2ml的Et2AlCl(0.5mol/L,甲苯中)以及剩余用来冲洗管道的30mL甲苯依次加入到250ml不锈钢高压釜中,使得甲苯总体积为100ml。此时Al/Ni=300:1。机械搅拌开始,保持400转/分,当聚合温度达到30℃时,往反应釜中充入乙烯,聚合反应开始。在30℃下保持10atm的乙烯压强,搅拌30min。用5%盐酸酸化的乙醇溶液中和反应液,得到聚合物沉淀,用乙醇洗数次,真空烘干至恒重,称重得2.71g聚合物,聚合活性:2.71×106g/mol(Ni)h-1,聚合物Tm=101.6℃。聚合物Mw=3.43kg·mol-1,Mw/Mn=4.96。
b)基本同a),区别在于:主催化剂为Ni2,助催化剂用量为1.4ml的Et2AlCl(0.5mol/L,甲苯中),使Al/Ni=350:1;聚合温度为30℃。聚合活性:3.38×106g/mol(Ni)h-1,聚合物Tm=100.2℃。聚合物Mw=2.62kg·mol-1,Mw/Mn=3.83。
c)基本同a),区别在于:主催化剂为Ni2,助催化剂用量为1.6ml的Et2AlCl(0.5mol/L,甲苯中),使Al/Ni=400:1;聚合温度为30℃。聚合活性:6.66×106g/mol(Ni)h-1,聚合物Tm=99.4℃。聚合物Mw=2.45kg·mol-1,Mw/Mn=3.74。
d)基本同a),区别在于:主催化剂为Ni2,助催化剂用量为1.8ml的Et2AlCl(0.5mol/L,甲苯中),使Al/Ni=450:1;聚合温度为30℃。聚合活性:4.78×106g/mol(Ni)h-1,聚合物Tm=99.7℃。聚合物Mw=2.05kg·mol-1,Mw/Mn=2.12。
e)基本同a),区别在于:主催化剂为Ni2,助催化剂用量为1.6ml的Et2AlCl(0.5mol/L,甲苯中),使Al/Ni=400:1;聚合温度为20℃。聚合活性:5.38×106g/mol(Ni)h-1,聚合物Tm=103.3℃。聚合物Mw=6.37kg·mol-1,Mw/Mn=5.79。支化度为49/1000。
f)基本同a),区别在于:主催化剂为Ni2,助催化剂用量为1.6ml的Et2AlCl(0.5mol/L,甲苯中),使Al/Ni=400:1;聚合温度为40℃。聚合活性:4.51×106g/mol(Ni)h-1,聚合物Tm=97.1℃。聚合物Mw=2.09kg·mol-1,Mw/Mn=3.96。
g)基本同a),区别在于:主催化剂为Ni2,助催化剂用量为1.6ml的Et2AlCl(0.5mol/L,甲苯中),使Al/Ni=400:1;聚合温度为50℃。聚合活性: 1.77×106g/mol(Ni)h-1,聚合物Tm=98.3℃。聚合物Mw=1.94kg·mol-1,Mw/Mn=3.78。
实施例17、利用配合物Ni1及Et2AlCl联合催化加压下的乙烯聚合
基本同a),区别在于:主催化剂为Ni1,助催化剂用量为1.6ml的Et2AlCl(0.5mol/L,甲苯中),使Al/Ni=400:1;聚合温度为30℃。聚合活性:5.84×106g/mol(Ni)h-1,聚合物Tm=53.2℃。聚合物Mw=1.12kg·mol-1,Mw/Mn=2.07。
实施例18、利用配合物Ni3及Et2AlCl联合催化加压下的乙烯聚合
基本同a),区别在于:主催化剂为Ni3,助催化剂用量为1.6ml的Et2AlCl(0.5mol/L,甲苯中),使Al/Ni=400:1;聚合温度为30℃。聚合活性:5.79×106g/mol(Ni)h-1,聚合物Tm=46.2℃。聚合物Mw=0.87kg·mol-1,Mw/Mn=2.95。
实施例19、利用配合物Ni5及Et2AlCl联合催化加压下的乙烯聚合
基本同a),区别在于:主催化剂为Ni5,助催化剂用量为1.6ml的Et2AlCl(0.5mol/L,甲苯中),使Al/Ni=400:1;聚合温度为30℃。聚合活性:6.36×106g/mol(Ni)h-1,聚合物Tm=71.9℃。聚合物Mw=1.07kg·mol-1,Mw/Mn=2.03。
实施例20、利用配合物Ni6及Et2AlCl联合催化加压下的乙烯聚合
基本同a),区别在于:主催化剂为Ni6,助催化剂用量为1.6ml的Et2AlCl(0.5mol/L,甲苯中),使Al/Ni=400:1;聚合温度为30℃。聚合活性:6.42×106g/mol(Ni)h-1,聚合物Tm=63.8℃。聚合物Mw=1.13kg·mol-1,Mw/Mn=1.95。
实施例21、利用配合物Ni7及Et2AlCl联合催化加压下的乙烯聚合
基本同a),区别在于:主催化剂为Ni7,助催化剂用量为1.6ml的Et2AlCl(0.5mol/L,甲苯中),使Al/Ni=400:1;聚合温度为30℃。聚合活性:5.77×106g/mol(Ni)h-1,聚合物Tm=45.9℃。聚合物Mw=0.53kg·mol-1,Mw/Mn= 1.74。
对照例1
将50mL甲苯、20mL配合物Ni1(2.0μmol)的甲苯溶液以及甲苯依次加入到250mL的不锈钢高压釜中,使总体积为100mL。当聚合温度达到30℃时,往反应釜中充入乙烯,保持1MPa的乙烯压力,搅拌反应30min。反应结束后释放压力,反应体系无活性。
对照例2
将50mL甲苯、20mL配合物Ni2(2.0μmol)的甲苯溶液以及甲苯依次加入到250mL的不锈钢高压釜中,使总体积为100mL。当聚合温度达到30℃时,往反应釜中充入乙烯,保持1MPa的乙烯压力,搅拌反应30min。反应结束后释放压力,反应体系无活性。
对照例3
将50mL甲苯、20mL配合物Ni3(2.0μmol)的甲苯溶液以及甲苯依次加入到250mL的不锈钢高压釜中,使总体积为100mL。当聚合温度达到30℃时,往反应釜中充入乙烯,保持1MPa的乙烯压力,搅拌反应30min。反应结束后释放压力,反应体系无活性。
对照例4
将50mL甲苯、改性的甲基铝氧烷(MMAO)(15mmol)以及甲苯依次加入到250mL的不锈钢高压釜中,使总体积为100mL。当聚合温度达到30℃时,往反应釜中充入乙烯,保持1MPa的乙烯压力,搅拌反应30min。反应结束后释放压力,反应体系无活性。
对照例5
将50mL甲苯、氯化二乙基铝(DEAC)(1mmol)以及甲苯依次加入到250mL的不锈钢高压釜中,使总体积为100mL。当聚合温度达到30℃时,往反应釜中充入乙烯,保持1MPa的乙烯压力,搅拌反应30min。反应结束后释放压力,反应体系无活性。
由上述实施例及对照例可知,本发明的配合物和助催化剂组成的催化剂组合物,表现出良好的乙烯聚合活性。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种咪唑酸萘二甲酰亚胺基乙酯类化合物及其用途的制作方法与工艺
- 一种肉桂酸萘二甲酰亚胺酯类化合物及其用途的制作方法与工艺
- 1,4,5,8‑萘四甲酰基二酰亚胺衍生物及其用途的制作方法与工艺
- 1,8‑萘二甲酰亚胺基杂环偶氮分散染料及其制备方法与流程
- 一种罗丹明‑双吡啶甲基氨基萘酰亚胺荧光探针REPN及其制备方法和应用与流程
- 以萘酰亚胺为核的双通道铜离子探针及其制备方法和应用与流程
- 一类含2-氨基嘧啶的萘酰亚胺化合物,其制备方法及应用与流程
- 含有二苯甲基萘的氢化喹啉亚胺镍配合物催化剂及其制备方法与应用与流程
- 一种基于萘酰亚胺衍生物的比色探针及其制备方法与应用与流程
- 一类含有4‑哌嗪基‑1,8‑萘酰亚胺的荧光二向色性染料,其制备方法及应用与流程