一种含硼化合物的制备工艺方法与流程
本发明具体涉及一种含硼药物的制备工艺,以及该制备工艺在该含硼产品工业化生产中的应用。
背景技术:
:
硼酸类化合物是有机化合物中极其重要的一类化合物,由于其独特的结构特征,具备良好的生物活性和药理作用,被广泛应用于合成潜在的酶抑制剂和反馈控制药物转运聚合物。而α-氨基硼酸更被认为是丝氨酸蛋白酶抑制剂的药效团,这类抑制剂能够可逆性地占据酶和底物作用的空间,使蛋白水解酶不能与底物多聚蛋白结合而水解相应的肽键,因而被制成包括凝血酶,肽键内切酶和二肽基肽酶等多种蛋白酶的抑制剂(如化合物E)。而丝氨酸蛋白酶还能调节细胞内和细胞外的蛋白水解酶水平,同时又是一种自由基清除剂和许多细胞因子的携带者和调节剂,所以α-氨基硼酸类似物还具有抗AIDS和抗肿瘤的活性(如化合物A,B,C和D)。基于上述广泛的药理和生物活性,近年来,α-氨基硼酸引起了国内外生物化学家和药物化学家的广泛关注。含硼化合物中“碳硼”化学键不如“碳碳”化学键稳定,因此,含硼化合物的制备一直是化学家面临的重大挑战。
蛋白酶体是泛素-蛋白酶体系统的重要组成部分,负责大多数细胞内蛋白质的调节和降解,蛋白质先被泛素(多肽)标记,然后被蛋白酶体识别和降解。蛋白酶体在调节细胞周期、细胞增殖和细胞凋亡方面起着核心作用。第一代蛋白酶体抑制剂硼替佐米(Bortezomib)等已经应用于临床多发性骨髓瘤治疗,具有抑制多种肿瘤细胞生长及放疗增敏作用。Ixazomib(MLN2238)是基于硼替佐米基础上的第二代蛋白酶体抑制剂。
MLN2238的合成制备一直是重大的科学挑战,MLN2238的纯化精制问题是该产品生产成本控制的难点。一般的高温条件下,碳硼化学键容易断裂,使得MLN2238的纯化工艺中尽量避免采用高温溶解重结晶的方法,这样给MLN2238的纯化提出了更大的挑战,如何尽量避免采用重结晶手段纯化MLN2238,一直困扰着化学家。
专利CN102066386中指出了化合物V的制备方法:
以上专利CN102066386采用中间体VII和中间体VIII进行偶联反应得到化合物VI,未经纯化直接投入下一步反应,在混合溶剂(甲醇/己烷)中,采用2-甲基丙硼酸进行硼酸酯交换脱保护得到化合物V的油状粗品,然后,采用极性较小的正庚烷反相析晶的方法制备得到化合物V的结晶,显然该方法无法去除制备工艺中引入的众多杂质。
以上方法存在诸多的缺点:
1、油状粘稠状中间体VI极难纯化,反应副产物杂质极难去除。中间体VI制备过程中的酰胺偶联试剂(例如:TBTU等)很难去除,造成制备得到的中间体化合物VI为油状粘稠状混合物,该中间体无法采用重结晶或者“常温打浆”的工艺进行纯化。
2、化合物V难纯化,反应副产物杂质极难去除。专利CN102066386没有公开化合物V的纯化方法,仅是化合物V从油状产物转变为结晶态产物的方法,该方法无法去除化合物V制备工艺中引入的杂质。经分析,产品化合物V中可能存在的工艺杂质分析如图1;
经分析可知,产品V中按照专利CN102066386中的合成工艺,可能存在以上7种合成工艺杂质,以上杂质的去除是化合物V制备的技术难点,但是专利CN102066386并没有公布纯化化合物V的具体工艺。专利WO2012177835中揭示了化合物II稳定性强于化合物V,但是该专利文献没有揭示性质稳定的化合物II如何转变为化合物I,专利WO2012177835中,仅是引用了专利CN102066386的工艺来制备化合物V。
综上所述,现有公开文献中,并没有揭示产品MLN2238的纯化策略,因此,开发和寻找适合工业化生产制备MLN2238的工艺和纯化方案,一直是医药工业界需要克服的科学难题。
技术实现要素:
:
现有公开文献中,并没有揭示产品MLN2238的纯化策略,本发明公开一种制备和纯化MLN2238(化合物V)的工艺路线,本条工艺路线首先在产品MLN2238制备完成前,设计了一种可以纯化的关键节点,该关键节点是性质更加稳定的固体化合物II。化合物II的性质已经在专利WO2012177835中进行了揭示,但是文献中化合物II的该性质并未见用于化合物V的制备纯化策略。所以本发明目的在于揭示将性质更加稳定的化合物II用于化合物V的合成。
本发明所要解决的技术问题是为了克服现有技术制备MLN2238(化合物V)的工艺路线的缺点,提供一种新型的制备MLN2238(化合物V)新方法。
本发明公开一种制备化合物V的方法:
该方法包括两个步骤:
步骤1,在一定的反应温度条件下,一定的溶剂中,通式化合物IV和通式化合物III反应得到通式化合物II;
步骤2,通式化合物II,在一定的温度条件下,一定的溶剂中,与酸或者碱反应得到化合物V;
其中,R1,R2独立的为C1~C6的烷基或氢,或者R1和R2连接构成烷烃环状结构;R3,R4独立的为C1~C6的烷基或氢,或者R3和R4分别为羰基;R5为C1~C6的烷基或氢。
本发明所述的制备化合物V的方法,其中R1,R2独立的为C1~C6的烷基或氢,或者R1和R2连接构成烷烃环状结构,选自:
R3,R4独立的为甲基或氢,或者R3和R4分别为羰基;R5为甲基或氢。
本发明所述的化合物V的制备方法,步骤1,其特征在于所述的反应溶媒选自醚类,C1~C8醇类,C3~C8酮类,C1~C8酯类,C5~C8烷烃类,C1~C8卤代烷烃类,芳香烷烃类中的一种或多种,步骤1,其特征在于所述反应温度为-10~80℃。
本发明所述的化合物V的制备方法,步骤1,其特征在于所述的反应溶媒优先选自:叔丁基甲基醚,乙醚,四氢呋喃,二氧六环,二氯甲烷,三氯甲烷,四氯化碳,乙腈,二氯乙烷,正己烷,正戊烷,正庚烷,乙醇,甲醇,叔丁醇,正丁醇,丙酮,丁酮,乙酸乙酯,甲苯,苯,二甲苯中的一种或多种,步骤1,其特征在于所述反应温度为0~70℃。
本发明所述的化合物V的制备方法,步骤2,其特征在于所述的反应溶媒选自醚类,C1~C8醇类,C3~C8酮类,C1~C8酯类,C5~C8烷烃类,C1~C8卤代烷烃类,芳香烷烃类中的一种或多种;步骤2,其特征在于所述反应温度为-10~80℃;步骤2,其特征在于所述酸为有机酸或者无机酸,所述碱为有机碱或者无机碱。
本发明所述的化合物V的制备方法,步骤2,其特征在于所述的反应溶媒2优先选自:叔丁基甲基醚,乙醚,四氢呋喃,二氧六环,二氯甲烷,三氯甲烷,四氯化碳,乙腈,二氯乙烷,正己烷,正戊烷,正庚烷,乙醇,甲醇,叔丁醇,正丁醇,丙酮,丁酮,乙酸乙酯,甲苯,苯,二甲苯中的一种或多种;步骤1,其特征在于所述反应温度为0~70℃;步骤2,其特征在于所述酸为盐酸,硫酸,磷酸,硝酸或者氯化氢的有机溶剂,或者硫酸,磷酸,硝酸与有机溶剂组成的混合酸溶液,所述碱为氢氧化钠,氢氧化钾。
本发明所述的化合物V的制备方法,
该方法包括两个步骤:
步骤1,在10~60℃反应温度条件下,乙酸乙酯溶剂中,通式化合物IV和通式化合物III络合反应得到通式化合物II;
步骤2,在10~60℃反应温度条件下,甲醇溶剂中,采用盐酸甲醇溶液脱保护得到化合物I;
其中R1,R2独立的为C1~C6的烷基或氢,或者R1和R2连接构成烷烃环状结构,选自: R3,R4独立的为甲基或氢,或者R3和R4分别为羰基;R5为甲基或氢。
本发明所述的化合物V的制备方法,其中R1和R2连接构成烷烃环状结构,选自:R3,R4独立的为甲基或氢;R5为甲基或氢。
本发明所述的化合物V的制备方法,
该方法包括两个步骤:
步骤1,在10~60℃反应温度条件下,乙酸乙酯溶剂中,化合物IV-1和通式化合物III络合反应得到通式化合物II;
步骤2,在10~60℃反应温度条件下,甲醇溶剂中,采用盐酸甲醇溶液脱保护得到化合物I;
其中R3,R4独立的为甲基或氢;R5为甲基或氢。
本发明所述的化合物V的制备方法,
该方法包括两个步骤:
步骤1,在20~60℃反应温度条件下,乙酸乙酯溶剂中,化合物IV-2和通式化合物III络合反应得到通式化合物II;
步骤2,在20~60℃反应温度条件下,甲醇溶剂中,采用盐酸甲醇溶液脱保护得到化合物I;
其中R3,R4独立的为甲基或氢;R5为甲基或氢。
综上所述,与现有技术相比本发明的有益效果总结如下:
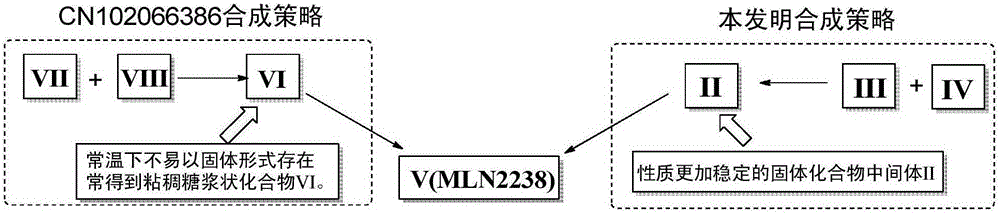
(1)、从工业化合成策略分析,本发明公开的制备化合物V的工艺路线中,采用性质稳定的固体化合物II作为质量控制的一个关键节点,有利于终产品化合物V(MLN2238)的质量控制。专利CN102066386工艺路线中,中间体VI由于偶联试剂很难与其分离,常常得到油状粘稠状混合物,该混合物包含了上文分析的7个潜在的工艺杂质,但是,化合物VI很难以固体形式存在,所以不能采用重结晶或者“打浆纯化”的方式除去以上潜在的7个工艺杂质,因此,化合物VI不利于作为质量控制的关键中间体节点。本发明与专利CN102066386合成策略比较如图2分析。
化合物VI若按照专利文献CN102066386方法制备,在制备过程中,由于后处理很难去除酰胺偶联试剂(如TBTU等偶联试剂),该化合物很难在常温下以固体形式存在,所以,化合物VI很难作为常规质量控制的关键中间体节点。因此,制备化合物VI时,产生的潜在的7个工艺杂质,很容易被带入终产品V(MLN2238)中。本发明采用性质更加稳定的固体化合物中间体II作为质量控制的节点,采用重结晶或者“打浆纯化”的方法纯化该质量控制的节点化合物II,这样合成工艺中大部分工艺杂质可以在中间体II的纯化过程中去除,不会将杂质带入终产品化合物V(MLN2238)。
(2)、本发明提供了一种化学制备工艺,将化合物II经过简单的化学工艺制备得到高纯度化合物V。专利WO2012177835揭示了化合物II性质稳定等优势特点,但是并没有指出如何将化合物II转化为化合物V,本发明采用简单的酸碱温和反应条件,由性质稳定化合物II制备得到高纯度的化合物V,,经HPLC测定纯度>99%。
综上所述,本发明在MLN2238的合成工艺路线改进中取得了显著的结果,对生产设备没有特殊的要求,反应条件温和,所有的原料均为市售产品,该工艺路线适合工业化生产,相信其在降低MLN2238原料药价格方面会做出巨大的贡献,有利于MLN2238相关产品的价格的大幅度的降低,造福于广大的患者。
附图说明:
图1显示了MLN2238合成工艺中潜在工艺杂质分析
图2显示了本发明合成工艺和已有文献资料报道工艺路线比较分析
具体实施方式:
下面用实施例来进一步说明本发明,但本发明并不受其限制。
实施例1:4-(R,S)-(羧基甲基)-2-((R)-1-(2-(2,5-二氯苯甲酰氨基)乙酰氨基)-3-甲基丁基)-6-氧代-1,3,2-二氧杂硼烷-4-甲酸
步骤1:N-2,5-二氯苯甲酰-L-甘氨酸甲酯
将2,5-二氯苯甲酸(15.00g,78.5mM,1.0eq)、L-甘氨酸甲酯盐酸盐(9.86g,78.5mM,1.0eq)、TBTU(30.25g,94.2mM,1.2eq)和四氢呋喃(200mL)的混合物冷却到0℃,搅拌30min,开始滴加二异丙基乙胺(38.92mL,235.5mM,3.0eq)。滴加完毕后,缓慢升至室温,反应过夜。经TLC监测反应完全,浓缩反应液,并加入乙酸乙酯,分别用饱和碳酸氢钠水溶液、柠檬酸水溶液、水和饱和食盐水洗涤,无水硫酸钠干燥,过滤并浓缩得到N-2,5-二氯苯甲酰-L-甘氨酸甲酯粗品。
步骤2:N-2,5-二氯苯甲酰-L-甘氨酸
将步骤1中所得N-2,5-二氯苯甲酰-L-甘氨酸甲酯粗品溶于丙酮(60mL)中,冷却至0℃,滴加2N氢氧化锂水溶液(40mL),滴加完毕后,升至室温(20-30℃)搅拌过夜。经TLC监测反应完全,浓缩反应液,并加入水,用乙酸乙酯萃取三次,将水层用浓盐酸调pH至2-3左右,用乙酸乙酯萃取两次,有机层分别用水和饱和食盐水洗2次,无水硫酸钠干燥,过滤并浓缩得到N-2,5-二氯苯甲酰-L-甘氨酸14.0g,收率72%。1H NMR(400MHz,DMSO-d6):δ=12.71(s,1H),8.90(s,1H),7.57–7.55(m,2H),7.50–7.47(m,1H),3.93(d,J=10.5Hz,2H)。
步骤3:N-2,5-二氯苯甲酰-L-甘氨酸-L-亮氨酸硼酸频哪醇酯
参照文献CN102066386方法制备:将N-2,5-二氯苯甲酰-L-甘氨酸(14.0g,56.4mM,1.0eq)、(R)-3-甲基-1-频哪醇二酯硼基-1-丁基胺盐酸盐(14.1g,56.4mM,1.0eq)、TBTU(19.0g,59.2mM,1.05eq)和二氯甲烷(250mL)的混合物冷却到0℃,搅拌30min,开始滴加二异丙基乙胺(28.0mL,169.2mM,3.0eq)。滴加完毕后,缓慢升至室温,反应过夜。经TLC监测反应完全,分别用水、1%磷酸水溶液、饱和碳酸氢钠水溶液、水和饱和食盐水洗涤,无水硫酸钠干燥,过滤并浓缩至45mL。
步骤4:2,5-二氯-N-[2-([(1R)-3-甲基-1-([1,3,6,2]二氧杂硼烷-2-基)丁基]氨基)-2-氧代乙基]苯甲酰胺
往步骤3中所得浓缩液(45mL)加入乙酸乙酯(250mL)和二乙醇胺(5.9g,56.4mM,1.0eq),室温搅拌过夜,固体过滤,然后得到的固体采用乙酸乙酯进行打浆纯化处理,除去大量反应产生的工艺杂质,过滤得到白色固体,乙酸乙酯洗,干燥得到目标产物19g,收率79%。1H NMR(400MHz,MeOD):δ=7.88–7.57(m,1H),7.62–7.17(m,2H),4.01–3.85(m,4H),3.86–3.77(m,1H),3.74–3.65(m,1H),3.44–3.35(m,1H),3.31–3.20(m,2H),2.98–2.86(m,2H),2.84–2.77(m,1H),1.74–1.59(m,1H),1.59–1.48(m,1H),1.44–1.29(m,1H),1.00–0.85(m,6H);13C NMR(101MHz,MeOD):δ=169.40,167.59,136.97,132.65,131.21,130.94,129.21,128.72,62.71,62.57,50.83,50.37,42.88,39.18,24.92,22.93,20.30。MS(ESI),m/z:430.15(计算为C18H27BCl2N3O4:430.15([M+H]+))。
步骤5:化合物V的制备
向步骤4中所得产物(19g,44.2mM)、甲醇(90mL),冷却至0℃,往其混合悬浮液滴入2N HCl(27mL,54mM),滴毕后,让其自然升温25℃过夜,用饱和碳酸氢钠水溶液调pH至6左右,浓缩掉甲醇,加入水,用乙酸乙酯萃取两次,合并乙酸乙酯层,用饱和氯化钠水溶液洗两次,收集有机相,无水硫酸钠干燥,过滤,浓缩至小体积即有大量的固体产生,过滤固体即得到目标产物14.2g,收率89%,经HPLC 测定纯度>99%。1H NMR(400MHz,MeOD):δ=7.64–7.59(m,1H),7.50–7.46(m,2H),4.39–4.24(m,2H),2.74–2.57(m,1H),1.77–1.61(m,1H),1.45–1.30(m,2H),0.93(d,J=6.6Hz,6H)。
实施例2:
将N-2,5-二氯苯甲酰-L-甘氨酸-L-亮氨酸硼酸频哪醇酯IV-1(7.0g,15.8mM,1.0eq)溶于甲酸乙酯(85mL)和二异丙醇胺(2.1g,15.8mM,1.0eq),0℃搅拌过夜,固体过滤,然后得到的固体采用乙酸乙酯进行打浆纯化处理,除去大量反应产生的工艺杂质,乙酸乙酯洗,干燥得到目标产物5.3g,收率73%。1H NMR(400MHz,MeOD):δ=7.68–7.61(m,1H),7.54–7.47(m,2H),4.39–4.08(m,2H),4.08–3.82(m,2H),3.57–3.40(m,0.5H),3.30–3.08(m,1H),3.09–2.98(m,1H),2.92–2.71(m,1.5H),2.71–2.48(m,1H),1.75–1.48(m,2H),1.45–1.11(m,7H),1.02–0.85(m,6H);13C NMR(101MHz,MeOD):δ=169.37,167.48,136.98,132.64,131.24,130.96,129.24,128.82,70.51,57.85,42.87,38.80,24.93,23.01,20.20,19.66;MS(ESI),m/z:458.26(计算为C20H31BCl2N3O4:458.18([M+H]+))
向上述所得产物(5.3g,11.6mM)中加入甲醇(28mL),冷却至10℃,往其混合悬浮液滴入2N HCl(7mL,14mM),滴毕后,让其升温至50℃反应1h,用饱和碳酸氢钠水溶液调pH至6左右,浓缩掉甲醇,加入水,用乙酸乙酯萃取两次,合并乙酸乙酯层,用饱和氯化钠水溶液洗两次,收集有机相,无水硫酸钠干燥,过滤,浓缩至小体积即有大量的固体产生,抽滤即得到目标产物3.6g,收率85%,经HPLC测定纯度>99%。
实施例3:
将N-2,5-二氯苯甲酰-L-甘氨酸-L-亮氨酸硼酸频哪醇酯IV-1(7.0g,15.8mM,1.0eq)溶于甲苯和二甲亚砜混合溶剂(85:5)(50mL)和N-甲基亚氨二乙酸(2.32g,15.8mM,1.0eq),60℃搅拌过夜,固体过滤,然后得到的固体采用乙酸乙酯进行打浆纯化处理,除去大量反应产生的工艺杂质,,乙酸乙酯洗,干燥得到目标产物4.3g,收率58%。
向上述所得产物(4.3g,9.1mM)中加入四氢呋喃(23mL),室温条件下,往其混合悬浮液滴入1NNaOH(6mL),滴毕后,让其自然升温25℃过夜,用饱和碳酸氢钠水溶液调pH至6左右,浓缩掉甲醇,加入水,用乙酸乙酯萃取两次,合并乙酸乙酯层,用饱和氯化钠水溶液洗两次,收集有机相,无水硫酸钠干燥,过滤,浓缩至小体积即有大量的固体产生,抽滤即得到目标产物2.9g,收率88%,经HPLC测定纯度>99%。
实施例4:
将N-2,5-二氯苯甲酰-L-甘氨酸(10.0g,40.3mM,1.0eq)、4-1(8.0g,40.3mM,1.0eq)(按照专利CN201210011233.2公开的方法制备)、TBTU(13.6g,42.3mM,1.05eq)和二氯甲烷(300mL)的混合物冷却到0℃,搅拌30min,开始滴加二异丙基乙胺(20.0mL,120.9mM,3.0eq)。滴加完毕后,缓慢升至室温,反应过夜。经TLC监测反应完全,分别用水、1%磷酸水溶液、饱和碳酸氢钠水溶液、水和饱和食盐水洗涤,无水硫酸钠干燥,过滤并浓缩完全。
往上述所得浓缩无中加入乙酸乙酯200mL,然后加入二乙醇胺(4.2g,40.3mM,1.0eq),室温25℃搅拌过夜,固体过滤,然后得到的固体采用乙酸乙酯进行打浆纯化处理,除去大量反应产生的工艺杂质,乙酸乙酯洗,干燥得到目标产物13g,收率75%。
向上述所得产物(13g,30.2mM)中加入乙醇(65mL),冷却至0℃,往其混合悬浮液滴入稀硝酸(3mL),滴毕后,让其0℃反应过夜,用饱和碳酸氢钠水溶液调pH至6左右,浓缩掉甲醇,加入水,用乙酸乙酯萃取两次,合并乙酸乙酯层,用饱和氯化钠水溶液洗两次,收集有机相,无水硫酸钠干燥,过滤,浓缩至小体积即有大量的固体产生,抽滤即得到目标产物9.4g,收率86%,经HPLC测定纯度>99%。
实施例5:
将N-2,5-二氯苯甲酰-L-甘氨酸(10.0g,40.3mM,1.0eq)、5-1(8.6g,40.3mM,1.0eq)(按照专利CN201210011233.2公开的方法制备)、TBTU(13.6g,42.3mM,1.05eq)和甲苯(200mL)的混合物冷却到0℃,搅拌30min,开始滴加二异丙基乙胺(59.9mL,362.7mM,3.0eq)。滴加完毕后,缓慢升至室温,反应过夜。经TLC监测反应完全,分别用水、1%磷酸水溶液、饱和碳酸氢钠水溶液、水和饱和食盐水洗涤,无水硫酸钠干燥,过滤并浓缩至150mL。
往上述所得浓缩液中加入二乙醇胺(4.2g,40.3mM,1.0eq),室温25℃搅拌过夜,固体过滤,然后得到的固体采用乙酸乙酯进行打浆纯化处理,除去大量反应产生的工艺杂质,乙酸乙酯洗,干燥得到目标产物12.6g,收率73%。
向上述所得产物(12.6g,29.3mM)中加入甲醇(66mL),冷却至0℃,往其混合悬浮液滴入1%浓硫酸乙醚溶液(18mL),滴毕后,让其自然升温25℃过夜,用饱和碳酸氢钠水溶液调pH至6左右,浓缩掉甲醇,加入水,用乙酸乙酯萃取两次,合并乙酸乙酯层,用饱和氯化钠水溶液洗两次,收集有机相,无 水硫酸钠干燥,过滤,浓缩至小体积即有大量的固体产生,抽滤即得到目标产物9.3g,收率88%,经HPLC测定纯度>99%。
实施例6:
将N-2,5-二氯苯甲酰-L-甘氨酸(8.0g,32.3mM,1.0eq)、6-1(8.6g,32.3mM,1.0eq)(按照专利CN200710088725.0公开的方法制备)、TBTU(10.9g,33.9mM,1.05eq)和二氯甲烷(150mL)的混合物冷却到0℃,搅拌30min,开始滴加二异丙基乙胺(21.4mL,129.2mM,3.0eq)。滴加完毕后,缓慢升至室温,反应过夜。经TLC监测反应完全,分别用水、1%磷酸水溶液、饱和碳酸氢钠水溶液、水和饱和食盐水洗涤,无水硫酸钠干燥,过滤并浓缩至100mL。
往上述所得浓缩液中加入二乙醇胺(3.4g,32.3mM,1.0eq),室温25℃搅拌过夜,固体过滤,然后得到的固体采用乙酸乙酯进行打浆纯化处理,除去大量反应产生的工艺杂质,乙酸乙酯洗,干燥得到目标产物9.9g,收率71%。
向上述所得产物(9.9g,23.0mM)中加入甲醇(55mL),冷却至0℃,往其混合悬浮液滴入2N HCl(14mL,28mM),滴毕后,让其自然升温25℃过夜,用饱和碳酸氢钠水溶液调pH至6左右,浓缩掉甲醇,加入水,用乙酸乙酯萃取两次,合并乙酸乙酯层,用饱和氯化钠水溶液洗两次,收集有机相,无水硫酸钠干燥,过滤,浓缩至小体积即有大量的固体产生,抽滤即得到目标产物6.8g,收率82%,经HPLC测定纯度>99%。
实施例7:
将N-2,5-二氯苯甲酰-L-甘氨酸(8.0g,32.3mM,1.0eq)、7-1(7.3g,32.3mM,1.0eq)(按照专利CN200710088725.0公开的方法制备)、TBTU(10.9g,33.9mM,1.05eq)和乙酸乙酯(150mL)的混合物冷却到0℃,搅拌30min,开始滴加二异丙基乙胺(21.4mL,129.2mM,3.0eq)。滴加完毕后,缓慢升至室温,反应过夜。经TLC监测反应完全,分别用水、1%磷酸水溶液、饱和碳酸氢钠水溶液、水和饱和食盐水洗涤,无水硫酸钠干燥,过滤并浓缩至100mL。
往上述所得浓缩液中加入二乙醇胺(3.4g,32.3mM,1.0eq),室温25℃搅拌过夜,固体过滤,然后得 到的固体采用乙酸乙酯进行打浆纯化处理,除去大量反应产生的工艺杂质,乙酸乙酯洗,干燥得到目标产物10.6g,收率76%。
向上述所得产物(10.6g,24.6mM)中加入甲醇(55mL),冷却至0℃,往其混合悬浮液滴入2N HCl(15mL,30mM),滴毕后,让其自然升温25℃过夜,用饱和碳酸氢钠水溶液调pH至6左右,浓缩掉甲醇,加入水,用乙酸乙酯萃取两次,合并乙酸乙酯层,用饱和氯化钠水溶液洗两次,收集有机相,无水硫酸钠干燥,过滤,浓缩至小体积即有大量的固体产生,抽滤即得到目标产物7.5g,收率84%,经HPLC测定纯度>99%。
实施例8:
将N-2,5-二氯苯甲酰-L-甘氨酸(12.0g,48.4mM,1.0eq)、8-1(15.6g,48.4mM,1.0eq)(按照专利CN201310258650.1公开的方法制备)、TBTU(16.3g,50.8mM,1.05eq)和二氯甲烷(200mL)的混合物冷却到0℃,搅拌30min,开始滴加二异丙基乙胺(24.0mL,145.2mM,3.0eq)。滴加完毕后,缓慢升至室温,反应过夜。经TLC监测反应完全,分别用水、1%磷酸水溶液、饱和碳酸氢钠水溶液、水和饱和食盐水洗涤,无水硫酸钠干燥,过滤并浓缩至150mL。
往上述所得浓缩液中加入二异丙醇胺(6.4g,48.4mM,1.0eq),室温25℃搅拌过夜,固体过滤,然后得到的固体采用乙酸乙酯进行打浆纯化处理,除去大量反应产生的工艺杂质,乙酸乙酯洗,干燥得到目标产物15.7g,收率71%。
向上述所得产物(15.7g,34.3mM)中加入甲醇(80mL),冷却至0℃,往其混合悬浮液滴入2N HCl(21mL,42mM),滴毕后,让其自然升温25℃过夜,用饱和碳酸氢钠水溶液调pH至6左右,浓缩掉甲醇,加入水,用乙酸乙酯萃取两次,合并乙酸乙酯层,用饱和氯化钠水溶液洗两次,收集有机相,无水硫酸钠干燥,过滤,浓缩至小体积即有大量的固体产生,抽滤即得到目标产物9.9g,收率80%,经HPLC测定纯度>99%。
虽然已通过上述具体实施方式和实施例详细说明了本发明的多个方面和不同实施方案,但本领域技术人员基于上述教导,将很容易预见到本发明所述方法、反应条件可具有适当的变化和调整,以适应具体的需要和实际情况,并且这些变化和调整均认为在本发明的范围内,即权利要求所限定的范围内。
- 还没有人留言评论。精彩留言会获得点赞!