一种制备增粘剂共聚缔合物的方法与流程
本发明涉及石油钻井过程中钻井液用聚合物的制备方法,特别是涉及一种钻井液用耐高温增粘剂及其制备方法。
背景技术:
在钻井过程中,为了保证低固相下钻井液具有较高的粘度及良好的流变性,通常需要添加增粘剂来提高钻井液的粘度。钻井液增粘剂均为分子链很长的水溶性高分子聚合物,增粘剂除了能起到增粘作用,还往往兼做页岩抑制剂(包被剂)、降滤失剂及流型改进剂等。因此,使用增粘剂常有利于改善钻井液的流变性,也有利于井壁稳定。
钻井液增粘剂是钻井助剂中最为重要的一种,主要分为天然植物胶和合成高分子两大类,在保证钻井液安全施工、携砂带屑等方面具有重要作用。然而现有的钻井液增粘剂如黄原胶、80A51 等,都不能很好的满足高温施工需要,大部分增粘剂在高于150℃条件下增粘效果迅速下降,甚至完全消失。在抗盐方面,抗高浓度氯化钙同样也是钻井液领域亟待解决的问题,目前常用的增粘剂中几乎没有抗氯化钙浓度超过15%的聚合物,所以说目前常用的钻井液用增粘剂耐温性能和抗盐抗钙性能往往不能兼顾。
CN102372818A 和CN102464761A 公开的抗钙聚合物增粘剂,主要是通过向丙烯酰胺类聚合物中引入磺化基团或疏水基团而得,仅可以满足钙离子浓度不高于2000mg/L 的低温条件下的(小于100℃)使用需求。CN101955564A、CN103113518A和CN102127401A公开的耐高温增粘剂,增粘剂的耐高温性能有显著提升,耐温达到200℃以上;抗盐抗钙性能并未提及,但从所用的单体及制备方法推测,抗盐抗钙性能无明显改善。
技术实现要素:
针对现有技术的不足,本发明提供一种制备增粘剂共聚缔合物的方法。本发明方法得到的增粘剂耐温可达200℃,抗NaCl浓度达饱和、抗CaCl2浓度达20%。在高温老化后保持了较高的表观粘度,同时还具有较好的动切力。
本发明提供了一种制备增粘剂共聚缔合物的方法,所述共聚缔合物含有结构单元A、结构单元B和结构单元C,所述结构单元A为N,N-二甲基丙烯酰胺,所述结构单元B为两性离子单体N-甲基二烯丙基丙磺酸盐(MAPS),所述结构单元C为结构单元B与阳离子聚胺的离子缔合体,以所述共聚缔合物的总量为基准,所述结构单元A的含量为10-75重量%,优选为30-65重量%;所述两性离子单体的总含量为8-55重量%,优选为20-60重量%;所述阳离子聚胺的含量为1-50重量%,优选为3-40重量%;所述共聚缔合物在200℃老化16小时后的表观粘度为40-70mPa·s;所述制备方法包括如下步骤:
(1)将无机盐与水混合,配制2wt%~30wt%的无机盐溶液;
(2)将阳离子聚胺与水混合配制成阳离子聚胺水溶液,所述阳离子聚胺水溶液的质量浓度为0.5%~3%;
(3)将乳化剂加入白油中,在搅拌条件下通N2除氧0.5~1h,制得油相,其中乳化剂与白油的质量比为1:(5~10);
(4)称取N-甲基二烯丙基丙磺酸盐和N,N-二甲基丙烯酰胺,然后加入至步骤(1)中配制的无机盐溶液中,待充分溶解后得到水相,N-甲基二烯丙基丙磺酸盐和N,N-二甲基丙烯酰胺单体总质量浓度为20%~40%,并且按照以为N,N-二甲基丙烯酰胺、N-甲基二烯丙基丙磺酸盐和阳离子聚胺的总量为基准,为N,N-二甲基丙烯酰胺的用量为10-75重量%,优选为30-65重量%;N-甲基二烯丙基丙磺酸盐的用量为15-65重量%,优选为20-60重量%;
(5)将步骤(4)制得的水相滴加入步骤(3)制得的油相中,同时进行搅拌,然后加入引发剂,待充分溶解后通入N2除氧0.5~1h,同时升温至40~60℃,反应1~2h;
(6)将步骤(2)配制的阳离子聚胺水溶液加入到步骤(5)得到的反应产物中,混合均匀,充分乳化后同时升温至50~70℃,继续反应3~5h,最后得到产品,以N,N-二甲基丙烯酰胺、N-甲基二烯丙基丙磺酸盐和阳离子聚胺的总量为基准,阳离子聚胺的用量为1-50重量%,优选为3-40重量%。
本发明制备方法中,步骤(1)中所用乳化剂为op15与司盘80的复合乳化剂或吐温80与司盘80的复合乳化剂,op15或吐温80与司盘80的质量比为1:(9~11)。
本发明制备方法中,步骤(5)中所述的油相中的白油与水相中的水的质量比为(2~4):(3~5)。
本发明制备方法中,所述结构单元A N,N-二甲基丙烯酰胺的结构式为: 式(I),所述结构单元B两性离子单体N-甲基二烯丙基丙磺酸盐(MAPS)的结构式为: 式(II),所述结构单元C具有下式所示的结构:
式(III)
其中,虚线…表示离子缔合作用,X-为无机阴离子;R、R9、R9’、R10、R10’各自为下述式(IV)所示的结构:式(IV),其中,R11为H、取代或未取代的C1-C5烷基,t为1-5的整数,z为0-5的整数;
n、p各自为1-5的整数;x为0-10的整数,y为1-10的整数;l、l’和l”的取值使得所述阳离子聚胺的运动粘度为100-500mm2/s,阳离子度为0.5-2mmol/g。进一步优选所述阳离子聚胺的运动粘度为150-450mm2/s,阳离子度为0.5-1.5mmol/g。
上述阳离子聚胺可以为各种具有多个氮正离子和相应的平衡负离子,优选情况下,所述阳离子聚胺由式(V)所示的端胺与式(VI)所示的环醚和式(VII)所示的卤代环氧烷通过缩合反应得到
t、n、p各自为1-5的整数;x为0-10的整数,y为1-10的整数。当x为0时,式(V)表示端二胺,y优选为1-7的整数;当x不等于0,且y=2时,式(V)表示多乙烯多胺,x为1-10的整数。
其中取代基和下角标的定义与前文相同。
具体地,所述阳离子聚胺可以通过下述方法制得:在搅拌条件及50-120℃下向端胺中滴加环醚,环醚和端胺的摩尔比为2-4:1,滴加结束后反应1-4小时,然后升温至80-150℃,搅拌条件下滴加卤代环氧烷,卤代环氧烷与端胺的摩尔比为0.2-0.7:1,滴加结束后反应1-4小时,然后终止反应。
需要说明的是,尽管升温前的温度范围50-120℃与升温后的温度范围80-150℃有部分重叠,但后者的温度必须比前者的温度高。
可以通过加入盐酸来终止反应。盐酸的加入量优选为盐酸:端胺=1-3:1(摩尔比)。
优选情况下,所述端胺为乙二胺、丙二胺、丁二胺、己二胺、二乙烯三胺、三乙烯四胺、四乙烯五胺中的一种或多种。
优选情况下,所述环醚为环氧乙烷、环氧丙烷、四氢呋喃中的一种或多种。
优选情况下,所述卤代环氧烷为环氧氯丙烷、环氧溴丙烷、环氧氯丁烷中的一种或多种。
本发明制备方法中,共聚缔合物是指聚合物中既包括通过共聚形成的结构,也包括通过离子缔合作用形成的结构,其中结构单元A、结构单元B通过共聚形成,结构单元B和阳离子聚胺通过离子缔合作用结合在一起,形成结构单元C。各结构式中的虚线…表示离子缔合作用。
本发明制备方法中,共聚缔合物中阳离子聚胺的含量是指共聚缔合物中由阳离子聚胺提供的各种结构形式的总含量,包括形成共聚缔合物的阳离子聚胺的量,也包括未形成共聚缔合物的阳离子聚胺的量。
由于发明人掌握的测试手段有限和/或基于现有测试手段的局限,本发明的共聚缔合物各结构单元的含量仅能测试为与单体对应的结构单元的含量,而不能测出结构单元C的含量,更不能测出参与缔合和未参与缔合的两性离子结构单元的量,即上述两性离子结构单元的含量包括与阳离子聚胺缔合形成结构单元C的两性离子结构单元即结构单元B的含量,也包括未与与阳离子聚胺缔合形成结构单元C的两性离子结构单元的含量。
本发明制备方法中,各个结构单元的含量可以通过反应前后单体含量计算或核磁共振结合红外光谱分析的方式来确定。
本发明制备方法中,所述C1-C5的烷基可以是甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、异戊基、叔戊基、新戊基中的一种或多种。所述C1-C5的烷基的取代基例如可以是卤素或羟基。
根据本发明提供的共聚缔合物,其中结构单元A、结构单元B之间为常规的共价聚合方式,即之间通过共价键结合,而结构单元C则通过其阴离子和阳离子分别与结构单元B上的阳离子和阴离子以离子缔合作用而成为共聚缔合物的结构单元,从而将其网络结构引入共聚缔合物中,使得共聚缔合物具有较高的切力和耐温耐盐性。上述离子缔合作用可以通过将其与通过将结构单元A、结构单元B之间的共聚缔合物与阳离子聚胺进行简单混合得到的混合物在相同条件下测得其切力和耐温耐盐性的巨大差异并结合化学原理推测得到。
本发明中,各个结构单元的含量可以通过反应前后单体含量计算或核磁共振结合红外光谱分析的方式来确定。
根据本发明提供的共聚缔合物的制备方法,所述无机盐的一个主要作用是提供两性离子结构单元上的阴离子和阳离子与阳离子聚胺上的阳离子和阴离子发生交换/缔合的环境,因此只要能够提供上述的环境的无机盐均可以用作本发明的无机盐。优选情况下,所述无机盐为铵盐、钙盐、镁盐、铜盐、锌盐、铝盐、锆盐中的一种或多种。所述无机盐优选以溶液形式使用。
本发明的发明人通过研究进一步发现,不同种类的无机盐,达到最佳效果的溶液浓度不同,例如,所述无机盐为铵盐时,所述无机盐溶液浓度优选为10wt%~30wt%;所述无机盐为钙盐、镁盐、铜盐、锌盐时,所述无机盐溶液浓度优选为5%~15%,更优选为10%~15%;所述无机盐为铝盐时,所述无机盐溶液浓度优选为2wt%~10wt%;所述无机盐为锆盐时,所述的无机盐溶液浓度优选为2wt%~5wt%。上述无机盐溶液的浓度仅考虑无机盐及其溶剂的量,不考虑单体等其他物质的量。
当所述无机盐为铵盐时,具体可以为氯化铵、溴化铵、硝酸铵的一种或多种;当所述无机盐为钙盐时,具体可以为氯化钙、溴化钙;当所述无机盐为镁盐时,具体可以为氯化镁、溴化镁、硫酸镁、硝酸镁的一种或多种;当所述无机盐为铝盐时,具体可以为氯化铝、溴化铝、硫酸铝、硝酸铝的一种或多种;当所述无机盐为铜盐时,具体可以为氯化铜、溴化铜、硫酸铜、硝酸铜的一种或多种;当所述无机盐为锌盐时,具体可以为氯化锌、溴化锌、硝酸锌的一种或多种;当所述无机盐为锆盐时,具体可以为氯化锆、溴化锆、氧氯化锆、硝酸锆的一种或多种。
本发明制备方法中,所述引发剂的种类和用量可以参照现有技术进行。优选情况下,所述引发剂可以为过硫酸钠、过硫酸钾、过硫酸铵中的一种或多种。所述引发剂的用量优选为单体总量的0.3-0.7wt%。
本发明方法中,所述单体总质量浓度为单体质量与水总质量的比值。
本发明制备方法中,所述阳离子聚胺具有下式(i)所示的结构:式(i)
其中,X-为无机阴离子;R、R9、R9’、R10、R10’各自为下述式(IV)所示的结构
式(IV),其中,R11为H、取代或未取代的C1-C5烷基,t为1-5的整数,z为0-5的整数;n、p各自为1-5的整数,x为0-10的整数,y为1-10的整数;l、l’和l’’的取值使得所述阳离子聚胺的运动粘度为100-500mm2/s,阳离子度为0.5-2mmol/g。
上述阳离子聚胺可以参照CN103773332A公开的阳离子聚胺聚合物的制备方法进行制备:通过将端胺、环醚和环氧卤代烷进行聚合反应得到。所述端胺可以为烷基二胺如碳原子数为1-6的烷基二胺,具体如乙二胺、丙二胺、丁二胺、戊二胺或己二胺;也可以为多烯多胺如NH2-(CH2-CH2-NH)nH其中n为1-5的整数,如二乙烯三胺、三乙烯四胺或四乙烯五胺。所述环醚可以为碳原子数为2-6的环醚如环氧乙烷、环氧丙烷、环氧丁烷。所述环氧卤代烷例如可以为环氧氯丙烷、环氧溴丙烷或环氧氯丁烷。但为了确保缔合作用和获得更好的钻井液性能,所述阳离子聚胺的运动粘度应当控制为100-500mm2/s,阳离子度应当控制为0.5-2mmol/g。可以通过控制环醚和环氧卤代烷的加入量来控制运动粘度和阳离子度在上述范围内。一般地控制端胺与环醚的摩尔比为1:(2-4)。端胺与环氧卤代烷的摩尔比为1:(0.2-0.7)。其他反应条件和操作可以参照上述现有技术进行。
所述阳离子聚胺优选以水溶液形式使用,阳离子聚胺水溶液的浓度优选为0.5-3wt%,其中水的量为反应体系中的总水量。
根据本发明提供的方法,所得产物用丙酮洗涤的目的在于除去未反应的组分,干燥的温度可以为100-120℃,干燥的时间可以为16-24小时。
本发明方法中,所用MAPS可按以下所述步骤合成:分别称取摩尔比为2:1~9:1,优选为2.5:1~8:1的N-甲基二烯丙基胺和1,3-丙磺酸内酯,并向N-甲基二烯丙基胺中加入1,3-丙磺酸内酯;然后在20℃~90℃的温度下反应0.5-4h,优选在60~80℃的温度下反应1-3h,最后经过滤、抽提、干燥制得N-甲基二烯丙基丙磺酸盐。所述1,3-丙磺酸内酯可以滴加到N-甲基二烯丙基胺中,也可以直接一次性加入到N-甲基二烯丙基胺中,优选直接一次性加入方式。当采用直接一次性加入方式时,N-甲基二烯丙基胺和1,3-丙磺酸内酯摩尔比优选为5.2:1~7.8:1;当采用滴加方式时,N-甲基二烯丙基胺和1,3-丙磺酸内酯摩尔比优选为2.5:1~5:1,滴加前可以将1,3-丙磺酸内酯加热熔化。所述抽提溶剂选用甲醇或乙醇,优选乙醇,抽提时间为1~3h,干燥温度为30~50℃,干燥时间为10~20h。
与现有技术相比,本发明提供的增粘剂共聚缔合物及其制备方法优点如下:
(1)本发明提供的共聚缔合物,通过以一定比例同时含有N,N-二甲基丙烯酰胺结构单元、两性离子结构单元和阳离子聚胺结构单元,且阳离子聚胺结构单元具有特定的运动粘度和阳离子度,使得该共聚缔合物用作钻井液增粘剂时,所得钻井液不仅在高温老化后具有良好的表观粘度,而且还具有较好的切力,且耐温至少可达200℃以上,抗NaCl浓度达饱和、抗CaCl2浓度达20%。
(2)本发明增粘剂共聚缔合物的制备方法中,在反应前期通过使用高浓度无机盐溶液,可以使磺酸基两性离子单体与无机盐结合形成特殊结构,由于无机盐中金属离子的存在使得磺酸基两性离子单体的结构更加伸展,聚合时明显降低分子间的空间位阻,使单体分子排列更加紧密,大大增加了聚合物的分子量。在反应后期通过加入低分子量阳离子聚胺,通过低分子量阳离子聚胺结构上的铵基正离子进一步与两性离子单体中的磺酸基发生作用,形成更加稳定的网络结构,加强了聚合物的结构粘度,在保证增粘剂具有良好的表观粘度的同时,又具有了较好的切力。
(3)本发明方法制备的增粘剂共聚缔合物具有长支链和刚性环状基团。在水溶液中由于长支链和刚性环状基团的存在,增加了聚合物的空间位阻,增大了聚合物的流体力学体积,导致聚合物受温度影响断裂水解的趋势减少,从而提高了其耐温的性能,耐温至少可达180℃,有的已达到200℃。
附图说明
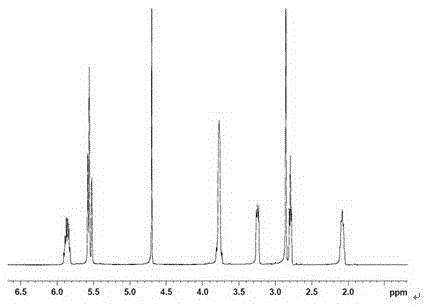
图1和图2分别为MAPS的1HNMR和13CNMR谱图。
具体实施方式
下面结合实施例来具体说明本发明方法的作用和效果,但以下实施例不构成对本发明方案的限制。下面的实施例将对本发明做进一步的说明。以下实施例中,运动粘度采用国标GB/T 265石油产品运动粘度测定法和动力粘度计算法测得;阳离子度采用胶体滴定方法测得;表观粘度及动切力由六速旋转粘度计方法测定。其中,切力表示体系具有的结构粘度,切力越大,说明钻井液的悬浮岩屑的性能越好。
本发明实施例和对比例中所用的MAPS单体可以按照如下方法制备:称取650g N-甲基二烯丙基胺倒入反应器中,然后放入恒温水浴锅中,加热并开始搅拌。再称取122g 1,3-丙磺酸内酯,直接加入到 N-甲基二烯丙基胺中,反应温度为80℃,搅拌反应3h后得到MAPS的粗产品。将MAPS粗产品转移到大片滤纸中包裹住,放置于索式抽提器中,使用乙醇为溶剂抽提3h,抽提完毕后将滤纸包放在干燥箱中,在50℃下干燥,最终得到纯净的MAPS单体。
本发明实施例和对比例中所用阳离子聚胺按照如下方法制备:
在1000mL四口圆底烧瓶中,加入60g乙二胺,在搅拌条件下升温至70℃,然后逐渐滴加116g环氧丙烷,控制反应温度60-100℃,反应1h后升温至95℃,逐渐滴加185g环氧氯丙烷,控制反应温度为90-150℃,待反应体系出现增稠现象时,至少维持半小时后加入盐酸,盐酸与乙二胺的摩尔比为2:1,反应4h得到阳离子聚胺1。阳离子聚胺1的运动粘度为330mm2/s,阳离子度为1.8mmol/g。
其中,R9、R9’、R10、R10’各自为下述式(IV-1)所示的结构式(IV-1)
在1000mL四口圆底烧瓶中,加入60g己二胺,在搅拌条件下升温至70℃,然后逐渐滴加116g环氧乙烷,控制反应温度60-100℃,反应1h后升温至95℃,逐渐滴加185g环氧溴丙烷,控制反应温度为90-150℃,待反应体系出现增稠现象时,至少维持半小时后加入盐酸,盐酸与己二胺的摩尔比为2:1,反应4h得到阳离子聚胺2。阳离子聚胺2的运动粘度为460mm2/s,阳离子度为1.9 mmol/g。
在1000mL四口圆底烧瓶中,加入60g三烯四胺,在搅拌条件下升温至70℃,然后逐渐滴加116g环氧丁烷,控制反应温度60-100℃,反应1h后升温至95℃,逐渐滴加185g环氧氯丁烷,控制反应温度为90-150,待反应体系出现增稠现象时,至少维持半小时后加入盐酸,盐酸与三烯四胺的摩尔比为2:1,反应4h得到阳离子聚胺3。阳离子聚胺3的运动粘度为410 mm2/s,阳离子度为1.5 mmol/g。
实施例1
将2.5gOP15和22.5g司盘80加入到150g白油中,在搅拌条件下通N2除氧半小时,制得油相备用;将37.5gNaCl加入到140g水中充分溶解后配制成NaCl水溶液,再将18gMAPS和63gDMAM加入到NaCl水溶液中,搅拌使之充分溶解;将油相转移至反应器中,搅拌的同时将水相滴加入油相中,搅拌半小时得到乳化体系;将0.24g过硫酸钾加入到乳化体系中,待充分溶解后通入N2除氧半小时,同时升温至60℃,反应1h后得到中间产物;再将2g阳离子聚胺1溶解于10g去离子水中配制成阳离子聚胺溶液,然后将阳离子聚胺溶液加入至中间产物中,混合均匀,同时升温至70℃,继续反应4小时。产物经破乳后,再用丙酮溶液洗涤,洗涤后的固体沉淀物在120℃干燥12h并粉碎后即为增粘剂。
实施例2
将1.5gOP15和13.5g司盘80加入到75g白油中,在搅拌条件下通N2除氧半小时,制得油相备用;将20gCaCl2加入到140g水中充分溶解后配制成CaCl2水溶液,再将47gMAPS和46gDMAM加入到CaCl2水溶液中,搅拌使之充分溶解;将油相转移至反应器中,搅拌的同时将水相滴加入油相中,搅拌半小时得到乳化体系;将0.32g过硫酸钠加入到乳化体系中,待充分溶解后通入N2除氧半小时,同时升温至55℃反应1.5h后得到中间产物;再将4g阳离子聚胺2溶解于10g去离子水中配制成阳离子聚胺溶液,然后将阳离子聚胺溶液加入至中间产物中,混合均匀,同时升温至65℃,继续反应3.5小时。产物经破乳后,再用丙酮溶液洗涤,洗涤后的固体沉淀物在110℃干燥12h并粉碎后即为增粘剂。
实施例3
将1.4g吐温80和13.6g司盘80加入到90g白油中,在搅拌条件下通N2除氧半小时,制得油相备用;将10gAlCl3加入到130g水中充分溶解后配制成AlCl3水溶液,再将28gMAPS和28gDMAM加入到AlCl3水溶液中,搅拌使之充分溶解;将油相转移至反应器中,搅拌的同时将水相滴加入油相中,搅拌半小时得到乳化体系;将0.2g过硫酸钾加入到乳化体系中,待充分溶解后通入N2除氧半小时,同时升温至50℃,反应2h后得到中间产物;再将6g阳离子聚胺3溶解于20g去离子水中配制成阳离子聚胺溶液,然后将阳离子聚胺溶液加入至中间产物中,混合均匀,同时升温至60℃,继续反应5小时。产物经破乳后,再用丙酮溶液洗涤,洗涤后的固体沉淀物在110℃干燥16h并粉碎后即为增粘剂。
实施例4
将1.8g吐温80和18.2g司盘80加入到120g白油中,在搅拌条件下通N2除氧半小时,制得油相备用;将5gZrCl4加入到140g水中充分溶解后配制成ZrCl4水溶液,再将14gMAPS和48gDMAM加入到ZrCl4水溶液中,搅拌使之充分溶解;将油相转移至反应器中,搅拌的同时将水相滴加入油相中,搅拌半小时得到乳化体系;将0.3g过硫酸钾加入到乳化体系中,待充分溶解后通入N2除氧半小时,同时升温至60℃,反应1h后得到中间产物;再将4g阳离子聚胺2溶解于10g去离子水中配制成阳离子聚胺溶液,然后将阳离子聚胺溶液加入至中间产物中,混合均匀,同时升温至65℃,继续反应4小时。产物经破乳后,再用丙酮溶液洗涤,洗涤后的固体沉淀物在120℃干燥12h并粉碎后即为增粘剂。
对比例1(不加无机盐)
将1.5gOP15和13.5g司盘80加入到75g白油中,在搅拌条件下通N2除氧半小时,制得油相备用;将47gMAPS和46gDMAM加入到140g水中,搅拌使之充分溶解;将油相转移至反应器中,搅拌的同时将水相滴加入油相中,搅拌半小时得到乳化体系;将0.32g过硫酸钠加入到乳化体系中,待充分溶解后通入N2除氧半小时,同时升温至55℃,反应1h后得到中间产物;再将4g阳离子聚胺2溶解于10g去离子水中配制成阳离子聚胺溶液,然后将阳离子聚胺溶液加入至中间产物中,混合均匀,同时升温至65℃,继续反应3.5小时。产物经破乳后,再用丙酮溶液洗涤,洗涤后的固体沉淀物在110℃干燥12h并粉碎后即为增粘剂。
对比例2(不加阳离子聚胺)
将1.5gOP15和13.5g司盘80加入到75g白油中,在搅拌条件下通N2除氧半小时,制得油相备用;将20gCaCl2加入到150g水中充分溶解后配制成CaCl2水溶液,再将47gMAPS和46gDMAM加入到CaCl2水溶液中,搅拌使之充分溶解;将油相转移至反应器中,搅拌的同时将水相滴加入油相中,搅拌半小时得到乳化体系;将0.32g过硫酸钠加入到乳化体系中,待充分溶解后通入N2除氧半小时,同时升温至65℃,反应5h后得到增粘剂乳液聚合物;产物经破乳后,再用丙酮溶液洗涤,洗涤后的固体沉淀物在110℃干燥12h并粉碎后即为增粘剂。
对比例3
通过市售购买增粘剂80A51。
对比例4
通过市售购买增粘剂黄原胶。
上述实施例及比较例使用含盐含钙的基浆评价增粘性能,具体评价方法如下:
基浆配制:在1000mL水中加入40g钙膨润土和5g碳酸钠,高速搅拌20min,室温下放置养护24h,得到淡水基浆;继续加入200g CaCl2,高速搅拌20min,室温下放置养护24h,得到评价用含20% CaCl2基浆。
评价方法:量取350mL的基浆,加入2%的增粘剂,高速搅拌20min,常温养护24h后测其表观粘度。再在200℃下老化16h后,再次测定表观粘度,计算表观粘度保持率。
表1 不同增粘剂耐温抗盐抗钙性能对比表
注:对比例4黄原胶的加量为0.5%
从表1 的结果可以看出,本发明的增粘剂在高温老化后表观粘度保持率较高,说明了较好的耐温及耐盐性能;并且高温老化后动切力还处于较好的水平,保证了钻井液体系在高温条件下的携岩提砂性能。
- 还没有人留言评论。精彩留言会获得点赞!