含反义碱基的探针法实时荧光PCR的制作方法
本发明属于分子生物学及分子检验领域的核酸扩增技术领域,具体涉及采用反义碱基的探针法实时荧光PCR来减少引物-探针聚合的非特异性的技术领域。
背景技术:
:
早在1953年,核酸DNA双螺旋模型及其中心法则开创了现代分子生物学及奠定了聚合酶链式反应(Polymerase Chain Reaction,PCR)的基础,甚至Khorana于1971年就直接提出了核酸体外扩增的设想:“经过DNA变性,与合适的引物杂交,用DNA聚合酶延伸引物,并不断重复该过程便可克隆tRNA基因”。但直到1983年,美国PE-Cetus公司人类遗传研究室的Kary Muliis在研究核酸测序技术时产生了指数扩增的灵感:于试管中模拟天然的DNA体内复制过程。提供一种合适的条件:模板DNA,寡核苷酸引物,DNA聚合酶,合适的缓冲体系,经过DNA变性---退火---延伸的循环过程,就可成几何级数地扩增已知两端序列的一段DNA分子几百万倍。经过一系列探索发展,耐热DNA聚合酶和热循环仪的发明与应用,Cetus公司于1987年申请了首个PCR发明专利(US Patent 4,683,202)。
PCR技术以其独有的特异性强、灵敏度高、简便快速和纯度要求低的特点而成为生命科学研究的核心基础技术。但传统的PCR(又称为终末PCR)需将产物凝胶电泳检测进行结果分析,传统的PCR由于PCR产物变异大的局限性,只能检测反应终末产物的定性而不能精确定量,同时也没有解决气雾胶污染、引物二聚体、非特异性扩增等难题,因此传统的PCR结合产物凝胶电泳检测方法不适用于临床检验等应用检测。1992年Higuchi等首次提出了采用动态PCR方法和封闭式荧光检测方式对目的基因数量进行分析,为解决传统PCR的难题提出了新思路。1996年由美国Applied Biosystems公司推出实时荧光定量PCR技术,所谓实时荧光定量PCR技术,是指在PCR反应体系中加入荧光基团,利用荧光信号积累实时监测整个PCR进程,最后通过标准曲线对靶模板进行定量分析的方法。扩增产物荧光信号达到设定的阈值所经历的循环数即Ct值(Cycle threshold)与靶模板起始拷贝数的对数存在负线性相关关系,即起始模板稀释一倍达到设定的阈值就要增加一个循环数(Ct)。该技术实行完全闭管式操作,避免了传统PCR开盖引起的产物污染问题,减少了假阳性结果的出现,同时也避免了对环境的污染。相比于传统PCR电泳的方法,该技术操作简便,检测结果以更客观的数据形式展现,从而对样本进行阳性、阴性和可疑的判定,保证了结果的准确性。
实时荧光定量PCR扩增产物增加的荧光信号既可通过荧光染料(如SYBR Green I)显示,又可采用带淬灭基团的荧光探针来检测。染料法即在PCR反应体系中,加入过量SYBR荧光染料,SYBR荧光染料非特异性地掺入DNA双链后,发射荧光信号,而不掺入链中的SYBR染料分子不会发射任何荧光信号,从而保证荧光信号的增加与PCR产物的增加完全同步。非特异的染料法成本较低,但面临着类似于传统PCR引物二聚体非特异性扩增产物干扰的假阳性问题。探针法即PCR扩增时在加入一对引物的同时加入一个特异性的荧光探针,该探针为一寡核苷酸,两端分别标记一个报告荧光基团和一个淬灭荧光基团。探针完整时,报告基团发射的荧光信号被淬灭基团吸收;PCR扩增时,Taq酶的5′-3′外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条 DNA链,就有一个荧光分子形成,实现了荧光信号的累积与PCR产物形成完全同步。与染料法相比,探针法在一定程度上避免了假阳性问题的出现,其特异性有引物和探针双重保证,进一步增加了结果的可信度。
1995年美国PE公司研制出了Taq酶水解的荧光标记探针及实时荧光定量PCR技术(Livak KJ,et al.,1995,Genome Res:4:357-362),于1997年申请了水解探针(商品名:TaqMan)PCR发明专利(US Patent 6,485,903);Epoch公司在水解荧光探针基础上研制了增加结合效率的MGB探针(US Patent 7,205,105),相较于TaqMan探针,该探针大大改善了本底信号的干扰,其较短的探针设计不但增强了淬灭效果,降低了荧光背景,同时能够较好地区分等位基因,可以检测单个碱基的突变(SNP)。2009年,美国的Igor V.Kutyavin提出了一种新的PCR检测技术-Snake系统,该系统通过在一条引物5’端添加一段与该条引物扩增产物探针5’端结合位点前面序列相同的特定碱基片段,使再一次扩增的产物自身3’端能够形成茎环状二级结构,再与探针结合后可作为Taq酶最佳的水解底物,增强了Taq酶5’→3’外切核酸酶的水解效率。不同于TaqMan,Snake系统可以使用较短的探针来改善荧光背景,在信号的产生和序列检测,尤其是单核苷酸检测方面有较强的优势。被淬灭的荧光探针也可以通过二级结构的改变而被激活,分子信标Molecular beacon(Tyagi S,et al,1996,Nat Biotechnol 14:303-308)正是这样一种茎环结构的杂交探针,于1999年申请了Molecular beacon发明专利(US Patent 05925517)。其它双杂交(FRET)探针,蝎形(Scorpion)探针,Sunrise-Primer,Lux Pimers等存在一定的应用局限性,不明显优于水解探针TaqMan方法,且与本发明路线、原理不同,这里不再赘述。目前临床检验使用最广的是水解探针TaqMan实时荧光PCR,而基础科研还是广泛使用基于荧光染料SYBR Green I的实时荧光PCR。
然而PCR巨大的放大倍数亦带来非特异性的巨大放大;且各种非特异性都远远赶不上一对引物的指数非特异性扩增,假阳性一直成为PCR应用中挥之不去的固有障碍;一般优化的PCR非特异性反应总在扩增30个循环时显著增加(Jannine Brownie,et al,1997,Nucleic Acids Research 25:3235-3241),这也是传统PCR只进行30个循环反应的原因。低浓度靶分子PCR与实时荧光定量PCR需要40个循环反应。SYBR Green I染料法实时荧光PCR虽解决了能定量问题,但其引物二聚体也结合染料,一般于Ct 30循环开始指数非特异扩增,假阳性遮盖了低浓度(低于5×105copies/反应)靶分子检测定量;不同于染料法,探针法使用的是只能与靶模板特异结合的标记有荧光基团和淬灭基团的一段寡核苷酸,绕过了引物二聚体产生荧光的假阳性问题。探针法PCR虽然引物二聚体不发射荧光信号,但引物二聚体非特异扩增竞争抑制低10倍以上浓度靶分子的同引物扩增(季新成等,2014,中国预防兽医学报36:646-650),低于34个循环的靶分子被PD抑制,牺牲了灵敏度,而不同引物的双重PCR的靶分子要相差100倍以上时才开始抑制(李杰等,2014,中华流行病毒学杂志6:720-723),所以引入与靶不同引物的非竞争性内标系统分散了靶引物的二聚体指数扩增而抵消了部分被PD抑制的假阴性(Pionzio AM,et al,2014,Forensic Sci Int Genet.9:55-60)。但内标增加了系统复杂性,同时也就增强了引物-探针聚合非特异性扩增,TaqMan实时荧光PCR不加靶模板的系统本底扩增一般于Ct 33-36开始爬升,尽管上升曲线及峰均很低,经几次重复PCR后,非特异性就开始严重起来了。
在实时荧光定量PCR检测中,为了确保反应液不蒸发和仪器光路通畅,多使用仪器热盖或矿物油封闭的策略(Sasaki N.1997,Hokkaido Igaku Zasshi.249-59)。由于实时荧光PCR“闭 管热盖”操作并不密闭,塑料的PCR反应管于热循环时软化,重复的蒸汽压力迫使大量气雾胶泄漏;既使反应液表面加矿物油后联合热盖,矿物油面上残留的反应液仍有效扩增、泄漏,只不过减轻了程度;热盖措施是由仪器工程师倡导应用的而不是生物学家促进使用的,热盖相较于单独使用矿物油密闭反而是一种退步。没了单独使用矿物油的密闭,探针法实时荧光PCR的引物-探针聚合的本底只要经3-7同样PCR的就立即出现本底扩增曲线明显提升,本底Ct从33-35前移至Ct 30甚至<Ct 30至Ct 25。
反义修饰寡核苷酸如甲基磷酸骨架或硫代磷酸骨架的修饰寡核苷酸,因能抵抗核酸酶水解而应用于基因沉默或敲除研究或作为抗癌药物;酰胺骨架的肽核酸(PNA)用于核酸杂交探针以增强结合与稳定性;锁核酸(LNA)探针实时荧光PCR提高了灵敏度,缩短了探针长度,相较于TaqMan探针法有更高的灵敏度、稳定性、扩增效率和较低的探针浓度,但目前也没有非特异性扩增的相关研究(边立忠等,2013,中华临床医师杂志7:2501-2506)(余文辉等,2011,中华生物医学工程杂志17:239-243)。为了彻底清除探针法实时荧光PCR的引物-探针间聚合扩增所产生的内源性非特异性反应,本发明“含反义碱基的探针法实时荧光PCR”于探针的中部序列引入1-2个或以上的2’-O-Methyl碱基,2’-Fluoro RNA碱基的改良探针,仍保留其5’端能被Taq酶5’外切酶活性所水解的能力,却失去作为与引物聚合的模板功能而不能被非特异性扩增。
技术实现要素:
:
为了消除TaqMan及MGB探针法实时荧光PCR仍有一些内生的引物-探针聚合非特异性扩增,采用探针序列中部引入1-2个反义碱基或以上而减少探针作为引物模板的可能性。
含反义碱基的探针法实时荧光PCR,其特征在于,所选的TaqMan及MGB探针离5’端5至20个碱基的中部序列中引入1至5个反义2’-O-Methyl碱基或2’-F-RNA碱基;其关键特征在于中部含反义碱基的寡核苷酸荧光TaqMan/MGB探针保留其5’端荧光基团标记的正常碱基序列仍能被Taq聚合酶5’外切酶活性所识别、水解;但探针失去了作为PCR扩增模板作用,引物非特异结合探针序列后Taq聚合酶不能识别反义碱基的模板而不能延伸、扩增,从而减少了内源性的引物-探针的聚合非特异性扩增。
所选的TaqMan/MGB探针离5’端10-15个碱基的中部序列中引入1至3个反义2’-O-Methyl碱基或2’-F-RNA碱基。
所述的改良探针法实时荧光PCR采用中部6-8个碱基同序的引物对以减少引物二聚体PD程度,从而减少对靶扩增的竞争性抑制,提高灵敏度。
所述的改良探针法实时荧光PCR探针3’末端添6-8个与其5’端反向互补的碱基序列,再标记淬灭基团,增加自身结合,而即降低荧光反应基线,又减少与引物结合的概率。
所述的PCR反应液加等体积的矿物油单独使用,封闭反应液,不联用热盖条件。
2.5U/ul Taq聚合酶加入0.1-2U/ul的UDG酶,消除PCR产物气雾胶的可能交叉污染。
所述的含反义碱基的探针法实时荧光PCR技术应用试剂盒,成份包括:核酸提取试剂,底物dNTPs,Taq聚合酶及其缓冲液,UDG酶,荧光探针,引物,去离子水。
不同于传统的检测信号相加的检验技术,其放大灵敏度有限。PCR以指数形式放大或几何倍数地扩增能放大百万倍以上而实现分子水平的检测,所以又被称为分子诊断。然而其最大缺点也源于非特异性的指数放大,虽然传统检测方法的非特异性也存在于PCR系统,但与 指数放大产生的非特异性相比可以忽略,引物与非特异DNA结合,或与另一引物3’端外的配对、延伸均是线性扩增,至多是两条线性扩增相加,均赶不上指数放大。只有一对引物3’末端均相互配对互补的引物二聚体PD扩增才是主要的指数形式的非特异性原因。一对3’末端存在2-3个互补碱基的引物,经过十几个热循环就会大量PD扩增而遮盖靶分子的扩增;经引物设计原则或软件优选的末端尽量无互补的引物对一般从30个循环开始PD指数扩增,仍限定了检测灵敏度。
水解探针TaqMan及MGB探针法实时荧光PCR由于探针不结合PD而绕过了PD产生的假阳性问题,但靶与PD扩增使用同引物的竞争性干扰作用十分严重,浓度较引物二聚体(Ct30)低十倍即对应Ct值在33之后的靶分子会被抑制产生假阴性(季新成等,2014,中国预防兽医学报36:646-650),对应的靶分子约为5×102copies/反应。同时根据Hands(Nucleic Acids Res.,Vol.25-16p3215)技术完全同序引物绝没有PD扩增,又根据中国专利(一对部分同序引物减少其二聚体的方法:中国,CN201010105371.8)部分同序引物部分减少PD程度,同序放在引物中部的关键部位能最大程度地减少PD。
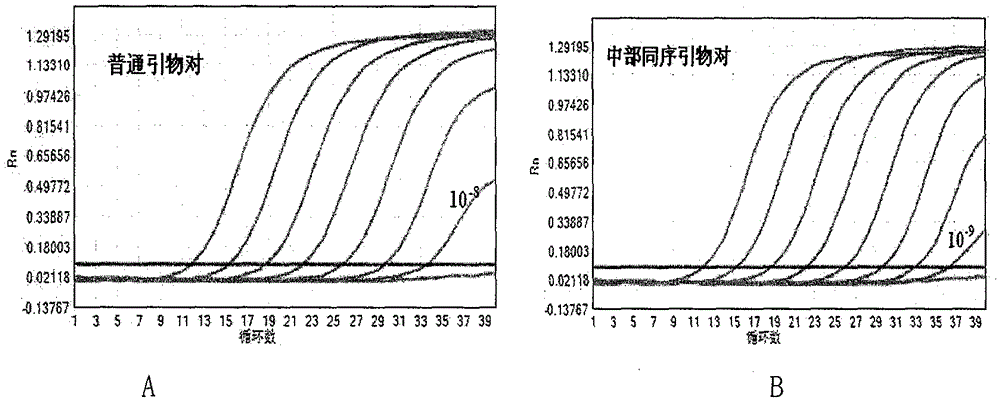
本专利采用SYBR Green I染料法检测普通引物对和中部同序引物对的PD扩增发现,普通引物对的Ct值在30左右,中部同序引物对的Ct值在38左右,中部同序引物对的PD非特异性扩增明显低于普通引物对(附图1)。采用探针法检测普通引物对和中部同序引物对结合探针检测系列稀释的β-globin模板,结果显示,普通引物对可以检测到10-8稀释度(如附图2A),中部同序引物对可以检测到10-9稀释度(如附图2B),TaqMan探针法中使用中部同序引物对检测灵敏度相比于普通引物对好一个数量级。HBV同序引物TaqMan结果灵敏度达Ct35-37,见实施例。
PCR被非特异性PD扩增造成靶抑制的假阴性往往被大多文献报道成由于样本中存在PCR抑制剂或提取方法不当引起的假阴性(Hidenobu Yaku,et al.2013,Molecules,11751-11767.),针对这一问题,大多采用引入内标系统的方式来解决(孟双.避免乙型肝炎病毒和丙型肝炎病毒核酸漏检的新型实时荧光PCR方法的研究及其内标的建立.北京协和医学院,2010.),即在靶分子扩增之外增加一内标对照基因的同时同管内扩增。与靶分子共用引物的竞争性内标因干扰靶扩增太强烈而一般不采用;非竞争性内标多采用管家基因,内标引物分散与推后靶引物指数扩增,由于不同引物的靶与内标双重PCR的模板要差100倍以上才部分影响抑制,高1000倍的内标才完全抑制靶分子扩增。
本专利选择HBV表面抗原质粒作为靶模板,HBV核心抗原质粒作为内标基因,二者使用不同引物对,进行干扰实验。结果见表6,当靶模版与内标模板浓度相差超过100倍时,基本没有影响或出现一些干扰,当二者浓度相差超过1000倍时,开始出现较严重的干扰作用(表1)。与上述文献基本一致。
表1 非竞争性内标与靶模板干扰实验
因此,必须选择经过样本提取之后Ct值在26-27之间的靶模板发挥内参照作用。Ct<17,或Ct>35的靶分子才会完全被抑制。所以采用中部同序引物提高灵敏度要好于内标方法。我们HBV采用中部同序引物联合内标β-globin系统。
尽管TaqMan及MGB探针法实时荧光PCR使用荧光探针绕开了PD的非特异性信号;但是引物对与探针之间仍存在一定的聚合非特异性扩增,其产物能结合并水解荧光TaqMan/MGB探针,有文献报道其假阳性率为4%-5%(裘宇容,等.2012,国际检验医学杂志,1365-1367),产生本底Ct值33-36左右的内源性假阳性扩增,尽管扩增曲线很低,但经过5-7次重复PCR反应后,扩增曲线显著增高,且本底Ct值逐渐前移。引入内标系统后增加了系统复杂性,非特异性假阳性更加明显。
本专利对比TaqMan探针、Molecular beacon、单独反义碱基探针和反义+beacon探针(探针序列见表2)的单重标准荧光PCR和加入内标系统的双重标准荧光PCR的本底假阳性累积实验,结果显示,单重PCR时,TaqMan探针和Molecular beacon开始即产生假阳性,并随着实验次数的增多逐渐加重;单独反义碱基探针和反义+beacon探针始终没有出现可检测到的假阳性;双重PCR时,TaqMan探针和Molecular beacon从第5次重复PCR反应开始即产生假阳性,并随着实验次数的增多逐渐加重,单独反义碱基探针和反义+beacon探针始终没有出现可检测到的假阳性,反义+beacon探针的基线相较于单独反义碱基探针低一些(表3、附图3)。反义碱基探针的性能明显好于TaqMan探针和Molecular beacon。
表2 探针序列
表3 假阳性积累结果
实时荧光PCR“闭管热盖操作”代替单独使用矿物油或联合使用矿物油均不密闭,热循环条件下塑料PCR管盖大量泄漏气雾胶;就是联合使用矿物油,加矿物油时溅起的油面上少量PCR反应液在热盖条件下仍有效扩增泄漏,只不过较单独“闭管热盖操作”的气雾胶程度要轻 一些而已;热盖较单独矿物油反而是一种退步。单独使用矿物油密闭是保证TaqMan PCR稳定可靠的前提保证。
本专利使用TaqMan探针在热盖、矿物油加热盖及矿物油无热盖三种条件下进行假阳性聚合实验,结果显示,矿物油无热盖(单独使用矿物油)PD严重程度低于其他两组(附图4)。
附图说明:
图1.为SYBR Green I染料法检测普通引物对和中部同序引物对的PD扩增,结果显示,普通引物对PD非特异性扩增的Ct值在30左右,中部同序引物对PD非特异性扩增的Ct值在38左右,中部同序引物对的PD非特异性扩增明显低于普通引物对。
图2.为普通引物对和中部同序引物对进行稀释的β-globin模板梯度实验,结果普通引物对可以检测到10-8稀释度,中部同序引物对可以检测到10-9稀释度,TaqMan探针法中使用中部同序引物对检测灵敏度相比于普通引物对好一个数量级。
图3.为TaqMan探针、Molecular beacon、单独反义碱基探针和反义+beacon探针的假阳性积累,结果单重PCR时,TaqMan探针和Molecular beacon开始即产生假阳性,并随着实验次数的增多逐渐加重;单独反义碱基探针和反义+beacon探针始终没有出现可检测到的假阳性;双重PCR时,TaqMan探针和Molecular beacon从第5次重复PCR反应开始即产生假阳性,并随着实验次数的增多逐渐加重,单独反义碱基探针和反义+beacon探针始终没有出现可检测到的假阳性。
图4.为TaqMan探针在热盖、矿物油加热盖及矿物油无热盖三种条件下进行假阳性聚合实验,结果热盖的假阳性随PCR重复次数增加而前移幅度最大,热盖加矿物油稍好于热盖,矿物油无热盖(单独矿物油)的假阳性PD严重程度均低于其他两组。
图5.为乙肝HBV改良TaqMan法实时荧光PCR标准品pHBcore 10倍稀释标准曲线,结果Ct值依次为15.5,19,23,26.5,29.3,31.5,35.3,本底对照Ct值在40循环内基本为一直线。对应的拷贝数为其3×109,3×108,3×107,3×106,3×105,3×104,3×103拷贝/ml,和本底0拷贝/ml。
图6.为乙肝HBV改良TaqMan法实时荧光PCR国家线性灵敏度参考品L0-L6 3次重复检测,结果线性灵敏度参考品L0-L5检出全部为阳性,定量浓度值也符合给出参考范围,相关系数R2不低于0.98。
图7.为人乙型肝炎病毒实时荧光TaqMan双重PCR检测标准品pHBcore 10倍稀释标准曲线,结果Ct值依次为15.4,19,22.8,26.6,29,32,35.3,本底对照Ct值在40循环内基本为一直线,对应的拷贝数为其3×109,3×108,3×107,3×106,3×105,3×104,3×103拷贝/ml,和本底0拷贝/ml。
图8.为乙肝HBV改良TaqMan法实时荧光PCR国家线性灵敏度参考品L0-L63次重复检测,结果线性灵敏度参考品L0-L5检出全部为阳性,定量浓度值也符合给出参考范围,相关系数R2不低于0.98。
具体实施例:
以下实施例进一步说明本发明的内容,但不应理解为对本发明的限制。以下实施例所用引物和探针均由生工生物工程(上海)股份有限公司合成,定量PCR仪为上海宏石SLAN-96P 实时荧光定量PCR仪。在不背离本发明精神和实质的情况下,对本发明方法、条件、步骤及应用所作的修改或替换,均属于本发明的范围。
实施例一:HBV改良TaqMan探针法实时荧光PCR:
探针法实时荧光PCR主要以TaqMan探针为代表,也包括增加结合力的MGB探针和锁核酸LNA碱基探针。扩增产物增加的信号通过带淬灭基团的荧光探针来检测。一般TaqMan探针5′端标记FAM、VIC、NED等荧光基团,3′末端标记TAMRA、DABCYL&BHQ等淬灭基团,被淬灭荧光的TaqMan探针通过Taq酶的5′外切酶水解而游离荧光基团产生荧光。TaqMan探针设计尊循以下一般原则:1)探针的Tm值比引物的Tm值要高10℃以上;2)探针5′端不能是G碱基,被酶切降解的G仍具有淬灭报告荧光作用;3)探针中的G不能多于C;4)避免单一核苷酸成串,尤其是G;5)富含AT的序列要增加长度,以达到符合要求的Tm值,但探针必须<40nt,否则淬灭效率低,反应本底高;6)探针退火时,其5′端应尽可能接近引物,同时又不重叠,离引物的3′端至少一个碱基远;7)检测单碱基变异(SNP)时,突变点尽量置于探针中间或靠近5′端,探针尽可能短;8)探针做mRNA表达分析时,探针序列应包括外含子/-/外含子边界;9)探针的3′端必须淬灭剂封闭以防止PCR扩增时延伸。
本发明采用改良TaqMan探针的实时荧光PCR反应,选取靶基因(临近下游一端引物)的一段代表性序列(nt:2306-2444),靶特异性序列(/互补序列)5’端标记报告荧光染料FAM,利用Taq酶5’-3’外切酶活性水解与靶序列杂交的探针而切割释放荧光基团;探针3’端标记淬灭基团BHQ-1,其探针中加入反义碱基;或整合TaqMan与Molecular beacon技术优点,探针3’端靶序列外添加几个与5’端互补碱基再标记淬灭基团BHQ-1,探针两末端类似Molecular beacon分子内末端互补形成锅柄样结构,不仅降低了探针与过量引物间形成非特异性杂交延伸的聚合体,也抑制降低了本底荧光2-4倍,兼容了Molecular beacon本底低和TaqMan灵敏、特异的优点。
本实例精选乙型肝炎病毒(HBV)核心Core区(CDR:2306-2444)一段序列作为HBV同序引物置换的核酸扩增靶特异序列,展示两端各20碱基Core区(nt:2306-2444)序列如下:
AB540584 Core区(nt:2306-2444)
CAAATGCCCC TATCTTATCA AC——GTCGCAGAAGA TCTCAATCTC
根椐同样一般引物选取原则以尽量减少引物二聚体来综合考虑、设计HBVcore探针法荧光PCR引物:
HBVc引物序列如下:
THBVFc:5′-ca aat gcc ccta tca ac-3′
THBVRc:5′-gag att gagc tgc gac-3′
其中F代表上游引物序列,R代表下游直接靶特异引物,粗体代表同序碱基。
根椐探针选取原则,设计HBVcore探针法荧光PCR探针:
TqHBc1:5′-FAM cgt ctg cga ggc/i2omeg/ag g/i2ome g/a gtt ctt ctt cta gc BHQ-1-3′
或TqHBc2:5′-FAM ctg cga ggc gag g/i2ome g/a gtt ctt ctt cgc ag BHQ-1-3′
(1)临床血标本DNA提取:
离心柱方法:取血清100μl,加100μl的沉淀液,涡流混匀,高速离心10分钟,弃上清,剩 余浓缩沉淀中加入50μl煮沸裂解液,轻混,置沸水或金属浴中煮沸10分钟,高速离心10分钟,上清即提取的DNA溶液。
微磁珠方法:采用盐酸胍/异硫氰酸胍裂解,核酸在高浓度4M胍盐条件下结合至聚苯乙烯微磁球硅烷化表面羟基(Melzak et al,1996),用小于pH6.0的缓冲液洗涤,大于pH8.5缓冲液洗脱。微磁球法已经越来越多地取代酚-氯仿抽提液体方法和硅吸附膜离心柱方法。
取血清100μl于1.5ml试管中,加等体积的胍盐裂解液5分钟,加0.8ml稀释中和液后再加25μl顺磁纳米硅化微球结合,将试管置于磁分离试管架吸附固定磁微球,弃液后,加0.8ml洗液洗涤一次,最后磁微球加50μl洗脱液收集DNA。
(2)TaqMan实时荧光PCR反应:
由于TaqMan探针PCR每个模板每次循环最多释放一个荧光基团,其反应荧光强度远低于SYBR Green I荧光染料法(每3-4bp DNA能结合一个SYBR Green I分子),所以TaqMan法必须采用较大体积的50μl反应体系,以使PCR仪器能接受到足够强度的荧光信号。
首先每个PCR反应管加2μl缓释引物R(2.5μM),缓释引物热启动和防产物气雾胶污染。再按以下配方取1.5ml的EP管配制不含待测模板和引物R的反应混合液,
(探针荧光会逐渐衰减,旧标记探针可相应多加一些。)
所配的×25次反应混合液共0.95ml分装于预加缓释引物的PCR反应管/或12管排管,每管38μl分装25管。其中一管加10μl纯化水dH2O作为PCR系统阴性对照,表面再小心、缓慢地沿管壁加上50μl矿物油封闭,短瞬离心,切记不要混匀!以防破坏缓释。
含2μl缓释引物R和38μl反应液的PCR管加10μl待检DNA,表面再小心、缓慢地加上50μl矿物油封闭。为了检测结果的可靠性,每次检测必须设立实验阳性阴性对照和定量校准品(根据是否定量需要)。
每个检测可平行2-3份×50μl计平均Ct值,分析统计结果。
标准曲线:
以pUC-HBcore(3.36kb,分子量MW=2.1×106,计1μg=2.8×1011copy分子)计算,相应标准品梯度分子拷贝数为:5.0×109/ml,5.0×108/ml,5.0×107/ml,5.0×106/ml,5.0×105/ml,5.0×104/ml和5.0×103/ml。
反应管上实时荧光PCR仪(调整仪器激发光波长FAM:480nm,检测光波长FAM:520nm)。按使用说明书设置运行程序,首先进行一个预反应50℃2分-94℃3分钟;然后PCR扩增40个热循环:94℃30秒,58℃30秒,72℃30秒;于72℃读取荧光值。
(3)实验结果分析:
HBV核心抗原质粒对照pUC-HBcore标准定量曲线的最左边第一条扩增曲线约为5.0×109 拷贝,随之作10倍稀释,最后一条扩增曲线为没有模板的本底对照,结果本底对照Ct值在40循环内基本为一直线,在PCR反应内几乎没有扩增Ct值(图5)。
购买中检所乙型肝炎病毒(HBV)核酸定量标准品(批号0711)阳性参考品及线性灵敏度参考品检测结果基本一致,阴性参考品全为基线反应。线性灵敏度参考品的灵敏度可达Ct35-37(图6)。临床试验500例阳性标本99%与上海克隆生物高技术有限公司检测结果符合,各种阴性血清检测未出现假阳性反应。多次重复性非常好。
实施例二:人乙型肝炎病毒实时荧光TaqMan双重PCR检测:
对于TaqMan探针法中的假阴性问题,现多采用加入内标组成双重PCR的方式,即在靶分子扩增之外增加一内标对照基因的同时同管内扩增。与靶分子共用引物的竞争性内标因干扰靶扩增太强烈而一般不采用;非竞争性内标多采用管家基因,内标引物分散与推后靶引物指数扩增,由于不同引物的靶与内标双重PCR的模板要差100倍以上才部分影响抑制,高1000倍的内标才完全抑制靶分子扩增。因此,必须选择经过样本提取之后Ct值在26-27之间的靶模板发挥内参照作用。靶Ct<17才抑制内标,或Ct>35的靶分子才会完全被抑制。我们HBV采用β-globin作为内标系统。
本发明采用改良TaqMan探针的实时荧光PCR反应,选取靶基因(临近下游一端引物)的一段代表性序列(nt:2306-2444),靶特异性序列(/互补序列)5’端标记报告荧光染料FAM,利用Taq酶5’-3’外切酶活性水解与靶序列杂交的探针而切割释放荧光基团;探针3’端标记淬灭基团BHQ-1,其探针中加入反义碱基;或整合TaqMan与Molecular beacon技术优点,探针3’端靶序列外添加几个与5’端互补碱基再标记淬灭基团BHQ-1,探针两末端类似Molecular beacon分子内末端互补形成锅柄样结构,不仅降低了探针与过量引物间形成非特异性杂交延伸的聚合体,也抑制降低了本底荧光2-4倍,兼容了Molecular beacon本底低和TaqMan灵敏、特异的优点。
本实例同样精选乙型肝炎病毒(HBV)核心Core区(CDR:2306-2444)一段序列作为HBV同序引物置换的核酸扩增靶特异序列,展示两端各20碱基Core区(nt:2306-2444)序列如下:
AB540584 Core区(nt:2306-2444)
CAAATGCCCC TATCTTATCA AC——GTCGCAGAAGA TCTCAATCTC
根椐同样一般引物选取原则以尽量减少引物二聚体来综合考虑、设计HBVcore探针法荧光PCR引物:
HBVc引物序列如下:
THBVFc:5′-ca aat gcc ccta tca ac-3′
THBVRc:5′-gag att gagc tgc gac-3′
其中F代表上游引物序列,R代表下游直接靶特异引物,粗体代表同序碱基。
根椐探针选取原则,设计HBVcore探针法荧光PCR探针:
TqHBc1:5′-FAM cgt ctg cga ggc /i2omeg/ag g/i2ome g/a gtt ctt ctt cta gc BHQ-1-3′
TqHBc2:5′-FAM ctg cga ggc gag g/i2ome g/a gtt ctt ctt cgc ag BHQ-1-3′
本发明采用改良TaqMan探针的实时荧光PCR反应,选取内标靶基因(临近下游一端引物) 的一段代表性序列(nt:308-488),靶特异性序列(/互补序列)5’端标记报告荧光染料HEX,利用Taq酶5’-3’外切酶活性水解与靶序列杂交的探针而切割释放荧光基团;探针3’端标记淬灭基团BHQ-1,其探针中加入反义碱基;或整合TaqMan与Molecular beacon技术优点,探针3’端靶序列外添加几个与5’端互补碱基再标记淬灭基团BHQ-1,探针两末端类似Molecular beacon分子内末端互补形成锅柄样结构,不仅降低了探针与过量引物间形成非特异性杂交延伸的聚合体,也抑制降低了本底荧光2-4倍,兼容了Molecular beacon本底低和TaqMan灵敏、特异的优点。
本实例精选人β-珠蛋白(Homo sapiens beta globin,HBB)一段序列作为β-globin同序引物置换的核酸扩增靶特异序列,展示两端各18碱基区(nt:308-488)序列如下:
KP309825 HBB(nt:308-488)
GTTTCTGATAGGCACTGACTC——GTGCTCGGTGCCTTTAGT
根椐同样一般引物选取原则以尽量减少引物二聚体来综合考虑、设计β-globin探针法荧光PCR引物:
β-globin引物序列如下:
βgF:5′-gtt tct gattga ctc-3′
βgR:5′-act aac gag cac-3′
其中F代表上游引物序列,R代表下游直接靶特异引物,粗体代表同序碱基。
根椐探针选取原则,设计β-globin探针法荧光PCR探针:
TqHBB:5′-HEX-ctt gcc atg agc ctt cac ctt ag-BHQ1-3′
(3)临床血标本DNA提取:
离心柱方法:取血清100μl,加100μl的沉淀液,涡流混匀,高速离心10分钟,弃上清,剩余浓缩沉淀中加入50μl煮沸裂解液,轻混,置沸水或金属浴中煮沸10分钟,高速离心10分钟,上清即提取的DNA溶液。
微磁珠方法:采用盐酸胍/异硫氰酸胍裂解,核酸在高浓度4M胍盐条件下结合至聚苯乙烯微磁球硅烷化表面羟基(Melzak et al,1996),用小于pH6.0的缓冲液洗涤,大于pH8.5缓冲液洗脱。微磁球法已经越来越多地取代酚-氯仿抽提液体方法和硅吸附膜离心柱方法。
取血清100μl于1.5ml试管中,加等体积的胍盐裂解液5分钟,加0.8ml稀释中和液后再加25μl顺磁纳米硅化微球结合,将试管置于磁分离试管架吸附固定磁微球,弃液后,加0.8ml洗液洗涤一次,最后磁微球加50μl洗脱液收集DNA。
(4)TaqMan实时荧光PCR反应:
由于TaqMan探针PCR每个模板每次循环最多释放一个荧光基团,其反应荧光强度远低于SYBR Green I荧光染料法(每3-4bp DNA能结合一个SYBR Green I分子),所以TaqMan法必须采用较大体积的50μl反应体系,以使PCR仪器能接受到足够强度的荧光信号。
首先每个PCR反应管加2μl缓释引物R(2.5μM),缓释引物热启动和防产物气雾胶污染。再按以下配方取1.5ml的EP管配制不含待测模板和引物R的反应混合液。
(探针荧光会逐渐衰减,旧标记探针可相应多加一些。)
所配的×25次反应混合液共0.95ml分装于预加缓释引物的PCR反应管/或12管排管,每管38μl分装25管。其中一管加10μl纯化水dH2O作为PCR系统阴性对照,表面再小心、缓慢地沿管壁加上50μl矿物油封闭,短瞬离心,切记不要混匀!以防破坏缓释。
含2μl缓释引物R和38μl反应液的PCR管加10μl待检DNA,表面再小心、缓慢地加上50μl矿物油封闭。为了检测结果的可靠性,每次检测必须设立实验阳性阴性对照和定量校准品(根据是否定量需要)。
每个检测可平行2-3份×50μl计平均Ct值,分析统计结果。
标准曲线:
以pUC-HBcore(3.36kb,分子量MW=2.1×106,计1μg=2.8×1011copy分子)计算,相应标准品梯度分子拷贝数为:5.0×109/ml,5.0×108/ml,5.0×107/ml,5.0×106/ml,5.0×105/ml,5.0×104/ml和5.0×103/ml。
反应管上实时荧光PCR仪(调整仪器激发光波长FAM:480nm,HEX:535nm,检测光波长FAM:520nm,HEX:553nm)。按使用说明书设置运行程序,首先进行一个预反应50℃2分-94℃3分钟;然后PCR扩增40个热循环:94℃30秒,58℃30秒,72℃30秒;于72℃读取荧光值。
(3)实验结果分析:
HBV核心抗原质粒对照pUC-HBcore标准定量曲线的最左边第一条扩增曲线约为5.0×109拷贝,随之作10倍稀释,最后一条扩增曲线为没有模板的本底对照,结果本底对照Ct值在40循环内基本为一直线,在PCR反应内几乎没有扩增Ct值(图7)。
购买中检所乙型肝炎病毒(HBV)核酸定量标准品(批号0711)阳性参考品及线性灵敏度参考品检测结果基本一致,阴性参考品全为基线反应。线性灵敏度参考品的灵敏度可达Ct35-37(图8)。临床试验500例阳性标本99%与上海克隆生物高技术有限公司检测结果符合,各种阴性血清检测未出现假阳性反应。多次重复性非常好。
- 还没有人留言评论。精彩留言会获得点赞!