高效sgRNA筛选系统和方法与流程
本发明涉及高效sgRNA筛选系统和方法。
背景技术:
现有的sgRNA活性检测是通过针对靶基因组序列设计多条sgRNA并克隆到表达载体(瞬时表达系统/慢病毒系统)中,再通过转染/慢病毒感染的方法导入目的细胞中,荧光蛋白(GFP)/嘌呤霉素(Puro)筛选的阳性细胞提取基因组后PCR扩增靶基因组序列进而通过错配酶(CEL1或T7E1酶)将识别错配的杂合双链剪切,产物DNA电泳后经过灰度分析评价sgRNA活性,电泳显示切割条带灰度越高活性越高。或者,直接将PCR扩增靶基因组序列进行深度测序(deep sequencing)分析出靶基因DNA序列出现缺失/插入(indel)的频率,频率越高则活性越高。或者直接将筛选的阳性细胞提取RNA/蛋白,直接检测靶基因的表达水平或蛋白水平以评价效率。前述的方法均需要长时间的细胞培养已及筛选操作,消耗大量的时间和试剂。错配酶分析常常出现假阳性切割且无法精确定量。深度测序昂贵并且需要分析大量测序数据。
技术实现要素:
本发明第一方面提供一种核酸构建物,该核酸构建物含有第一报告基因和位于第一报告基因3’端的第二报告基因。
在一个具体实施例中,所述第一报告基因编码荧光素酶蛋白或荧光蛋白。
在一个具体实施例中,所述第二报告基因编码荧光蛋白或荧光素酶蛋白。
在一个具体实施例中,所述第一报告基因表达的报告蛋白不同于所述第二报告基因表达的报告蛋白。
在一个具体实施例中,所述核酸构建物为pCAG-EGFP-Luc质粒。
在一个具体实施例中,所述核酸构建物的序列如SEQ ID NO:1所示。
在一个具体实施例中,所述核酸构建物还含有位于第一报告基因5’端的靶基因序列。
在一个具体实施例中,第一报告基因的起始密码子前含有一段多克隆位点(MCS)。
在一个具体实施例中,所述靶基因序列紧邻第一报告基因的5’端,且位于所述多克隆位点的上游。
在一个具体实施例中,所述靶基因序列紧邻第一报告基因5’端,或位于所述多克隆位点内部。
在一个具体实施例中,所述多克隆位点的DNA序列如SEQ ID NO:第591-665位碱基所示。
本发明第二方面提供一种核酸构建物,所述核酸构建物表达CRISPR-Cas9及识别所述靶基因序列的sgRNA。
在一个具体实施例中,所述核酸构建物采用lentiCRISPRv2质粒构建获得。
本发明第三方面提供一种产品及其在评价(如定性和/或定量)sgRNA介导的基因组修饰活性中的用途,该产品含有以下核酸构建物:
(1)含有第一报告基因的核酸构建物;和
(2)表达CRISPR-Cas9的核酸构建物。
在一个具体实施例中,(1)所述核酸构建物还含有靶基因序列,该靶基因序列位于第一报告基因的5’端;(2)所述核酸构建物还表达识别(1)所述靶基因序列的sgRNA。
在一个具体实施例中,(1)所述核酸构建物还含有位于第一报告基因的3’端的第二报告基因。
在一个具体实施例中,第一报告基因编码荧光素酶蛋白或荧光蛋白。
在一个具体实施例中,第二报告基因编码荧光蛋白或荧光素酶蛋白。
在一个具体实施例中,第一报告基因表达的报告蛋白不同于第二报告基因表达的报告蛋白。
在一个具体实施例中,(1)所述核酸构建物含有荧光素酶编码序列和位于荧光素酶编码序列5’端的荧光蛋白编码序列。
在一个具体实施例中,该产品为试剂盒。
在一个具体实施例中,所述试剂盒还含有用于将靶基因插入(1)所述构建物中第一报告基因的5’端的试剂,和/或用于在(2)所述核酸构建物中插入sgRNA的试剂。
在一个具体实施例中,所述试剂盒还含有用于将(1)和(2)所述核酸构建物转入宿主细胞的试剂。
在一个具体实施例中,所述试剂盒还含有用于检测荧光素酶酶活的试剂。
本发明第四方面提供以下所述核酸构建物在评价(如定性和/或定量)sgRNA介导的基因组修饰活性中的用途:
(1)含有第一报告基因的核酸构建物;和
(2)表达CRISPR-Cas9的核酸构建物。
在一个具体实施例中,(1)所述核酸构建物还含有靶基因序列,该靶基因序列位于第一报告基因的5’端;(2)所述核酸构建物还表达识别(1)所述靶基因序列的sgRNA。
在一个具体实施例中,(1)所述核酸构建物还含有位于第一报告基因的3’端的第二报告基因。
在一个具体实施例中,第一报告基因编码荧光素酶蛋白或荧光蛋白。
在一个具体实施例中,第二报告基因编码荧光蛋白或荧光素酶蛋白。
在一个具体实施例中,第一报告基因表达的报告蛋白不同于第二报告基因表达的报告蛋白。
在一个具体实施例中,(1)所述核酸构建物含有荧光素酶编码序列、位于荧光素酶编码序列5’端的荧光蛋白编码序列和位于荧光蛋白编码序列5’端的靶基因序列。
在一个具体实施例中,(1)所述核酸构建物为pCAG-EGFP-Luc质粒。
在一个具体实施例中,(1)所述核酸构建物的序列如SEQ ID NO:1所示。
在一个具体实施例中,所述靶基因为感兴趣的基因组基因或其部分。
在一个具体实施例中,靶基因的长度为40~500个碱基,优选40~100个碱基,更优选为40~80个碱基。
本发明第五方面提供一种评价(如定性和/或定量)sgRNA介导的基因组修饰活性的方法,或筛选具有较高的基因组修饰活性的sgRNA的方法,所述 方法包括:
(1)提供或构建含第一报告基因和位于第一报告基因5’端的靶基因序列的表达载体;
(2)提供或构建表达CRISPR-Cas9和sgRNA的表达载体,其中,所述sgRNA识别所述靶基因序列;
(3)将(1)和(2)所述表达载体共转染入宿主细胞中;和
(4)观察和/或检测报告基因的表达情况,从而评价所述sgRNA介导的基因组修饰活性。
在一个具体实施例中,所述宿主细胞是HEK293细胞。
在一个具体实施例中,(1)所述核酸构建物还含有位于第一报告基因的3’端的第二报告基因。
在一个具体实施例中,第一报告基因编码荧光素酶蛋白或荧光蛋白。
在一个具体实施例中,第二报告基因编码荧光蛋白或荧光素酶蛋白。
在一个具体实施例中,第一报告基因表达的报告蛋白不同于第二报告基因表达的报告蛋白。
在一个具体实施例中,(1)所述核酸构建物含有荧光素酶编码序列、位于荧光素酶编码序列5’端的荧光蛋白编码序列和位于荧光蛋白编码序列5’端的靶基因序列。
在一个具体实施例中,(1)所述核酸构建物为pCAG-EGFP-Luc质粒。
在一个具体实施例中,(1)所述核酸构建物的序列如SEQ ID NO:1所示。
在一个具体实施例中,所述靶基因为感兴趣的基因组基因或其部分。
在一个具体实施例中,靶基因的长度为40~500个碱基,优选40~100个碱基,更优选为40~80个碱基。
本发明第六方面涉及一种构建用于基因组编辑的慢病毒的方法,所述方法包括:
(1)提供或构建含第一报告基因和位于第一报告基因’端的靶基因序列的表达载体;
(2)提供或构建表达CRISPR-Cas9和sgRNA的慢病毒表达载体,其中,所述sgRNA识别所述靶基因序列;
(3)将(1)和(2)所述表达载体共转染入宿主细胞中;和
(4)观察和/或检测报告基因的表达情况,筛选获得具有所需sgRNA介导的基因组修饰活性的(2)所述的慢病毒表达载体;
(5)在宿主细胞中由步骤(4)所获得的慢病毒表达载体制备慢病毒。
在一个具体实施例中,(1)所述核酸构建物还含有位于第一报告基因的3’端的第二报告基因。
在一个具体实施例中,第一报告基因编码荧光素酶蛋白或荧光蛋白。
在一个具体实施例中,第二报告基因编码荧光蛋白或荧光素酶蛋白。
在一个具体实施例中,第一报告基因表达的报告蛋白不同于第二报告基因表达的报告蛋白。
在一个具体实施例中,(1)所述核酸构建物含有荧光素酶编码序列、位于荧光素酶编码序列5’端的荧光蛋白编码序列和位于荧光蛋白编码序列5’端的靶基因序列。
在一个具体实施例中,(1)所述核酸构建物为pCAG-EGFP-Luc质粒。
在一个具体实施例中,(1)所述核酸构建物的序列如SEQ ID NO:1所示。
在一个具体实施例中,所述靶基因为感兴趣的基因组基因或其部分。
在一个具体实施例中,靶基因的长度为40~500个碱基,优选40~100个碱基,更优选为40~80个碱基。
本发明的优点在于:利用本发明的质粒(尤其是同时含第一报告基因和第二报告基因的质粒,如pCAG-EGFP-Luc质粒)可以简单、快速构建任何靶基因的模拟基因组,不受细胞系或物种的限制。另外,本发明相比传统的sgRNA活性检测手段可以更快速、简便评价sgRNA活性,并且可以精确定量sgRNA活性。避免了人工、时间、试剂的浪费,为实现CRISPR-Cas9技术更高效的用于基因组编辑奠定基础。
附图说明
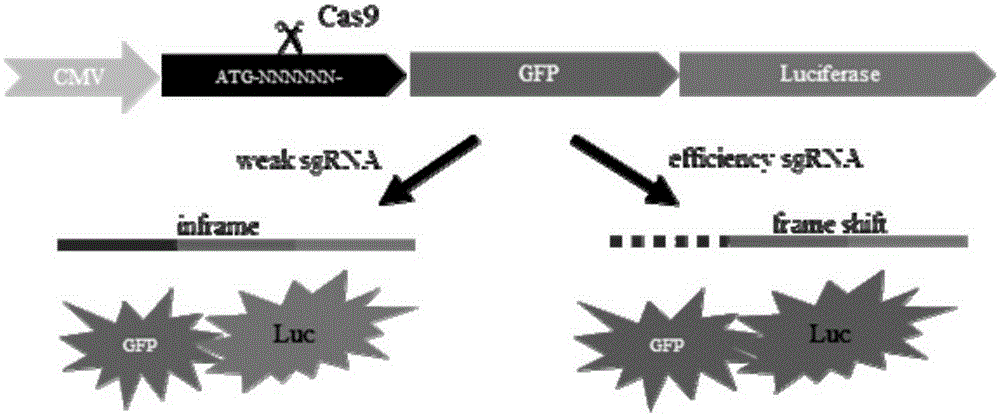
图1为本发明pCAG-EGFP-Luc质粒用于sgRNA编辑基因组活性测定的示意图。图中显示,高效的sgRNA将导致Cas9切割pCAG-EGFP-Luc后发生移码,从而沉默绿色荧光蛋白报告基因和荧光素酶报告基因表达的活性。
图2为本发明sgRNA编辑基因组活性测定系统检测靶向小鼠RIP1基因的两条sgRNA在HEK293T细胞中绿色荧光蛋白报告基因表达结果。采用本发明检测系统可以鉴定出2#sgRNA具有高效沉默绿色荧光蛋白报告基因表达的活性。图中,EV表示没有sgRNA。gmRIP1-1和gmRIP1-2是针对同一基因的不同sgRNA,靶向的基因组位置有所不同。gmRIP1-1和gmRIP1-2分别对应SEQ ID NO:4和5。
图3为本发明sgRNA编辑基因组活性测定系统检测靶向小鼠RIP1基因的两条sgRNA在HEK293T细胞中荧光素酶报告基因酶活力检测结果,本发明检测系统可以鉴定出2#sgRNA具有高效沉默荧光素酶报告基因表达的活性。
图4为本发明筛选获得的靶向小鼠RIP1基因的高效sgRNA利用慢病毒系统在小鼠L929细胞系中敲除RIP1基因的蛋白水平检测结果,2#sgRNA可以高效敲除小鼠L929细胞中RIP1蛋白水平。
图5为利用本发明筛选获得的靶向小鼠RIP1基因的高效sgRNA建立的小鼠L929细胞RIP1敲除细胞系诱导细胞坏死处理的检测结果,2#sgRNA敲除小鼠L929细胞中RIP1蛋白水平可以使细胞缺失RIP1的功能。图中,“Naive”表示原始细胞,不经过任何感染的细胞;“Moi”表示慢病毒感染系数,值越大病毒颗粒越多;“Ctrl”表示正常培养基加入药物空白溶解液DMSO;“T+Z”表示正常培养基加入药物TNFα及zVAD,诱导细胞坏死,细胞不能存活,只有RIP1沉默情况下细胞才能存活;“T+Z+N”表示正常培养基加入药物TNFα及zVAD,以及RIP1抑制剂Nec1,细胞坏死被抑制,细胞存活。
具体实施方式
本发明公开了一种用于快速评价sgRNA介导的基因组修饰活性的系统和载体。构建含第一报告基因(如荧光素酶全长编码序列)和/或第二报告基因(如绿色荧光蛋白编码序列)的质粒。将CRISPR-Cas9靶向的基因组序列插入第一或第二报告基因起始密码子前,将该质粒与sgRNA以及CRISPR-Cas9的表达质粒共同转染HEK293T细胞,通过观察第一报告基因表达的报告蛋白和/或第二报告基因表达的第二报告蛋白从而能快速定性/定量测定sgRNA介导的基因组修饰活性。该系统筛选获得的高效sgRNA可以用于CRISPR-Cas9慢病毒系 统建立靶基因敲除细胞系。
为实现本发明目的,本发明提供一种核酸构建物(在此称为核酸构建物A),该核酸构建物A用于表达第一报告基因编码的第一报告蛋白。通常,在第一报告基因的起始密码子之前还含有一段多克隆位点。
优选的是,核酸构建物A还含有编码第二报告蛋白的第二报告基因(本文称为核酸构建物A’),该第二报告基因通常位于第一报告基因的3’端。
报告蛋白可以是例如荧光素酶和荧光蛋白。荧光蛋白可以是,如绿色荧光蛋白、红移荧光蛋白和蓝色荧光蛋白。
第一报告基因和第二报告基因可表达相同的报告蛋白,也可表达不同的报告蛋白,优选表达不同的报告蛋白。例如,第一报告基因和第二报告基因分别表达荧光素酶和荧光蛋白,或者表达不同的荧光蛋白。
更具体而言,该核酸构建物A'含有:
(1)第一报告基因;
(2)位于第一报告基因3’端的第二报告基因;和
(3)位于第一报告基因起始密码子前的一段多克隆位点。
在本发明的一个具体实施例中,核酸构建物A’含有:
(1)荧光素酶编码序列;
(2)位于荧光素酶编码序列5’端的绿色荧光蛋白编码序列;和
(3)位于绿色荧光蛋白的起始密码子前的一段多克隆位点。
荧光素酶和绿色荧光蛋白及其编码序列都是本领域周知的。换言之,可使用本领域州的荧光素酶和绿色荧光蛋白的编码序列来构建本发明的核酸构建物A。
对于用于构建核酸构建物A的骨架载体也无特殊限制。例如,在一个具体实施例中,本发明以pEGFP-N1(Clontech)为起始载体,构建本发明的核酸构建物A。构建所得的核酸构建物A为pCAG-EGFP-Luc质粒。
在一个具体实施例中,所述核酸构建物A的序列如SEQ ID NO:1所示。
应用于本发明的方法中时,需要使所述核酸构建物A或A’含有CRISPR-Cas9靶向的序列,尤其是基因组序列。
因此,此时,所述核酸构建物A或A’还含有位于第一报告基因5’端的靶 基因序列。通常,所述靶基因序列紧邻第一报告基因的5’端,且第一报告基因的起始密码子前含有一段多克隆位点(MCS)。在一个具体实施例中,所述多克隆位点的DNA序列如SEQ ID NO:1的第591-665位碱基所示。
靶基因序列可以是任何感兴趣的基因组基因序列或其部分。通常并不需要将完整的基因序列插入本发明的核酸构建物A或A’中,而仅需要将其特异性的或保守的序列片段插入核酸构建物A或A’即可。例如,靶基因序列通过长40~500bp,更常见的长度为40~100bp。
为实现本发明的目的,本发明提供另一种核酸构建物(本文称为核酸构建物B),所述核酸构建物B表达CRISPR-Cas9及识别所述靶基因序列的sgRNA。可采用已知的骨架载体构建核酸构建物B。例如,在本发明的一个具体实施例中,所述核酸构建物采用lentiCRISPRv2质粒构建获得。核酸构建物B中sgRNA依靶基因的不同而不同。
核酸构建物A或A’和核酸构建物B可以独立的产品形式提供,也可以组合的形式提供。
优选的是,提供核酸构建物A或A’与核酸构建物B的组合(产品),例如提供于同一个试剂盒中。此时,核酸构建物A或A’可以是空载体,即不含靶基因序列的载体;同样地,核酸构建物B也可以是空载体,即不含sgRNA的载体。技术人员可根据实际待测的基因组基因而设计相应的靶基因序列,并将其插入到核酸构建物A或A’的第一报告基因的5’端。同时,根据所选的靶基因序列设计相应的sgRNA,并将其插入核酸构建物B中。应理解,sgRNA在核酸构建物B中的位置应能使得核酸构建物B能识别靶基因序列并允许Cas9切割所述靶基因序列。这对于本领域技术人员而言是显而易见的。
所述产品/试剂盒优选还可含有用于将靶基因插入核酸构建物A或A’中第一报告基因5’端的试剂,和/或用于在核酸构建物B中插入sgRNA的试剂。
用于将靶基因插入核酸构建物A或A’中第一报告基因5’端的试剂包括但不限于用于将核酸构建物A或A’线性化的各种酶,如Xho I和Hind III,用于将靶基因连接到线性化的核酸构建物A或A’的DNA连接酶,如T4DNA连接酶等。试剂还可包括用于构建所得核酸构建物A或A’转化大肠杆菌的试剂以及用于提取质粒的制剂等。
用于在核酸构建物B中插入sgRNA的试剂包括但不限于:将核酸构建物B线性化的各种酶,例如BsmB I;去磷酸化的酶,例如CIAP碱性磷酸酶;以及DNA连接酶,例如T4DNA连接酶。同样地,试剂还可包括用于构建所得核酸构建物A转化大肠杆菌的试剂以及用于提取质粒的制剂等。
除此之外,产品/试剂盒中还含有用于检测荧光素酶酶活的试剂。
应理解的是,上述核酸构建物A或A’、核酸构建物B以及将靶基因序列插入核酸构建物A或A’及将sgRNA插入核酸构建物B的技术手段都在本领域技术人员所掌握的范围之内。
本发明的核酸构建物A或A’和核酸构建物B,以及含有所述核酸构建物A或A’和核酸构建物B的产品可用于评价(如定性和/或定量)sgRNA介导的基因组修饰活性。
具体而言,针对插入核酸构建物A或A’的对应于感兴趣的基因组序列的靶基因序列设计多条sgRNA序列,然后将其插入核酸构建物B中。然后将两种核酸构建物共转染到宿主细胞如HEK293细胞中。通过观察和/或检测报告基因的表达情况或活力,即可判定对应的sgRNA介导的基因组修饰活性的强弱。
图1示例性阐述了本发明的原理。图中显示,高效的sgRNA将导致Cas9切割pCAG-EGFP-Luc,切割将发生移码,从而沉默绿色荧光蛋白报告基因和荧光素酶报告基因表达的活性。如sgRNA识别靶基因的特异性或效率不高或较弱,则Cas9将无法切割pCAG-EGFP-Luc或切割的数量不足,将导致不产生绿色荧光蛋白和荧光素酶或所产生的量较少。从而通过对比不同sgRNA产生的绿色荧光蛋白强度以及荧光素酶酶活,也可判定哪个sgRNA具有较强的基因组修饰活性。
可采用本领域常规的技术手段实施转染。例如,使用Lipofectamine2000(Invitrogen)进行转染。转染的其它技术手段可根据例如Lipofectamine2000(Invitrogen)的制造商提供的说明书进行。
可采用常规的技术手段检测报告蛋白的表达情况。不同报告蛋白其检测手段未必相同,但这都在本领域技术人员所掌握的范围之内。例如,可采用常规的技术手段检测绿色荧光蛋白及荧光素酶酶活力测定。例如,可在荧光显微镜下激光激发GFP并拍照记录,可采用荧光素酶报告基因检测试剂盒进行反应, 使反应物在光度计上测定相对光单位(RLU)。由此即可定性也可定量评价sgRNA的基因组修饰活性。
因此,本发明提供一种评价(如定性和/或定量)和/或sgRNA介导的基因组修饰活性的方法,所述方法包括:
(1)提供或构建本发明核酸构建物A或A’,其中,所述核酸构建物A或A’含位于第一报告基因5’端的靶基因序列;
(2)提供或构建表达CRISPR-Cas9和sgRNA的表达载体,其中,所述sgRNA识别所述靶基因序列;
(3)将(1)和(2)所述表达载体共转染入宿主细胞中;和
(4)观察和/或检测报告基因的表达或活性,从而评价所述sgRNA介导的基因组修饰活性。
如前所述,宿主细胞是HEK293细胞。
本发明还提供一种构建用于基因组编辑的慢病毒的方法,所述方法包括:
(1)提供或构建本发明核酸构建物A或A’,其中,所述核酸构建物A或A’含位于第一报告基因5’端的靶基因序列;
(2)提供或构建表达CRISPR-Cas9和sgRNA的慢病毒表达载体,其中,所述sgRNA识别所述靶基因序列;
(3)将(1)和(2)所述表达载体共转染入宿主细胞中;和
(4)观察和/或检测报告基因的表达或活性,筛选获得具有所需sgRNA介导的基因组修饰活性的(2)所述的慢病毒表达载体;
(5)在宿主细胞中由步骤(4)所获得的慢病毒表达载体制备慢病毒。
适用于本发明的慢病毒载体可以是本领域常用的任何慢病毒载体,包括但不限于市售载体,例如lentiCRISPRv2质粒(Addgene plasmid#52961)。
在宿主细胞如HEK293T细胞中由慢病毒表达载体制备慢病毒的方法也是本领域周知的,包括利用转染试剂如Lipofectamine2000(Invitrogen)将步骤(4)所获得的慢病毒表达载体转染进宿主细胞的步骤。同时转染的还包括例如适用慢病毒构建的其它载体,如psPAX2和VSV-G等。随后按常规技术手段培养所述宿主细胞,并收集病毒。
下文将以具体实施例的方式阐述本发明。应理解,这些实施例仅仅是阐述 性的,并非限制本发明。除非另有说明,否则实施例中所采用的方法、试剂和条件均为本领域常规的方法、试剂和条件。
实施例1、质粒pCAG-EGFP-Luc质粒的构建
pEGFP-N1(Clontech)质粒用Not I酶切线性化和CIAP碱性磷酸酶去磷酸化;以pGL3-Basic(Promega)质粒为模板PCR扩增荧光素酶全长CDS,PCR产物纯化后与线性化的pEGFP-N1进行拼装反应(Gibson Assembly,NEB)。热激转化大肠杆菌后涂布平板,挑取单克隆,测序确定序列完全正确,获得pCAG-EGFP-Luc质粒,序列如SEQ ID NO:1所示。
实施例2、pCAG-target-EGFP-Luc质粒的构建
2.1.人工合成正向单体DNA序列为(5’-TCGAGATGN1N2N3---N60-3’),以及反向单体DNA序列为(5’-TTCGA N'60---N'3N'2N'1CAT-3')。其中,N'x为Nx的反向互补序列,N和N'表示碱基A、T、G或C,N1-N60包含所有sgRNA识别的序列。
2.2.将上述合成的针对靶基因的两个单体DNA退火形成具有粘性末端的双链DNA。
2.3.实施例1构建得到的pCAG-EGFP-Luc质粒用Xho I及Hind III双酶切线性化后与双链DNA混合,T4 DNA连接酶连接后转化大肠杆菌感受态DH5α。提取质粒,即构建得到pCAG-target-EGFP-Luc质粒。
实施例3、通过筛选小鼠RIP1基因高效sgRNA验证sgRNA筛选系统的有效性
3.1.构建模拟小鼠RIP1基因组的荧光-荧光素酶双报告基因质粒——pCAG-mRIP1-EGFP-Luc质粒
3.1.1.人工合成正向单体DNA序列
(5’-TCGAGATGATGGCATCCAGTGACCTGCTGGAGAAGACAGACCTAGA CAGCGGAGGCTTCGGGA-3’,SEQ ID NO:2);和反向单体DNA序列(5’-TTCGATCCCGAAGCCTCCGCTGTCTAGGTCTGTCTTCTCCAGCAGG TCACTGGATGCCATCAT-3’,SEQ ID NO:3)。
3.1.2.将上述合成的针对靶基因的两个单体DNA退火形成具有粘性末端的双链DNA;
3.1.3.实施例1构建获得的pCAG-EGFP-Luc质粒用Xho I及Hind III双酶切线性化后与双链DNA混合,T4DNA连接酶连接后转化大肠杆菌感受态DH5α。提取质粒,即构建得到pCAG-mRIP1-EGFP-Luc质粒。
3.2.构建靶向小鼠RIP1的sgRNA载体——lentiCRISPRv2-sgRNA质粒
3.2.1.设计靶向mRIP1的sgRNA,选取sgRNA序列为
Target 1:5’-GATGGCATCCAGTGACCTGC-3’(SEQ ID NO:4)
Target 2:5’-TCCCGAAGCCTCCGCTGTCT-3’(SEQ ID NO:5)
3.2.2.将上述合成的针对同一个sgRNA作用位点的两个sgRNA单体退火形成具有粘性末端的双链DNA;
3.2.3.lentiCRISPRv2质粒(Addgene plasmid#52961)BsmB I线性化和CIAP碱性磷酸酶去磷酸化后与sgRNA双链DNA混合,T4DNA连接酶连接后转化大肠杆菌感受态DH5α。提取质粒,即构建得到lentiCRISPRv2-sgRNA质粒。
3.3.测定靶向小鼠RIP1sgRNA的活力
3.3.1.细胞株HEK293T的复苏与培养
取液氮冻存的HEK293T细胞,放于37℃水浴中解冻;解冻后800rpm条件下离心2min迅速加入1ml体积分数为10%胎牛血清的DMEM培养液中(DMEM培养液需要提前预热至37℃),枪头轻轻吹打至无细胞团存在;再将离心管中细胞悬液转移至8ml提前预热至37℃体积分数为10%胎牛血清的DMEM培养液的10cm培养皿中,均匀分布细胞后,于37℃、含5%的CO2培养箱中培养24h,以后每隔2天换液至细胞密度达90%时传代。
3.3.2.lentiCRISPRv2-sgRNA及pCAG-mRIP1-EGFP-Luc质粒共转染HEK293T
转染前一天,按照每孔1x105 HEK293T细胞接种到12孔板,培养24h后 细胞约有70%融合,转染前4h,将含血清的培养液换为无双抗无血清的opti-MEM(1ml/孔);Lipofectamine2000(Invitrogen)作为转染试剂将1ug前述构建获得的lentiCRISPRv2-sgRNA及2ug前述构建获得的pCAG-mRIP1-EGFP-Luc转染至HEK293T细胞,具体步骤依照说明书进行(所有试剂均为一个孔的试剂用量):用opti-MEM培养液将3ug无内毒素的质粒稀释至50ul,室温孵育;同样用opti-MEM培养液将3ul Lipofectamine2000稀释至50ul;将孵育的质粒和Lipofectamine2000混匀,总体积为100ul,混合溶液室温孵育30min;将100ul混合液加到培养板的一个孔中,轻轻晃动培养板混匀培养液;将细胞置于37℃、含5%的CO2培养箱中培养,6h后将培养液换为体积分数为10%胎牛血清的DMEM培养液继续培养18h。
3.3.3.荧光观察及荧光素酶酶活力测定
HEK293T细胞转染24h后更换含有2ug/ml嘌呤霉素培养基(体积分数为10%胎牛血清的DMEM培养液)中培养2d后倒置荧光显微镜(奥林巴斯)下激光激发GFP并拍照记录,结果如图2所示。
去除培养基后收集细胞按萤火虫荧光素酶报告基因检测试剂盒(碧云天)说明书操作,反应物在Luminometer(Thermo)测定RLU(relative light unit,相对光单位)。结果如图3所示。
实施例4、验证sgRNA筛选系统在慢病毒系统中的有效性
4.1.构建靶向小鼠RIP1的高效sgRNA的慢病毒载体
4.1.1.人工合成两个sgRNA单体,其序列分别为:
正向单体:5’-CACCGTCCCGAAGCCTCCGCTGTCT-3'(SEQ ID NO:6),
反向单体:5’-AACAGACAGCGGAGGCTTCGGGAC-3'(SEQ ID NO:7)。
4.1.2.将上述合成的针对同一个sgRNA作用位点的两个sgRNA单体退火形成具有粘性末端的双链DNA。
4.1.3.lentiCRISPRv2质粒(Addgene plasmid#52961)BsmB I线性化和CIAP碱性磷酸酶去磷酸化后与sgRNA双链DNA混合,T4DNA连接酶连接后转化大肠杆菌感受态DH5α。提取质粒,即构建得到lentiCRISPRv2-sgRNA质粒。
4.2.lentiCRISPRv2-sgRNA慢病毒制备
4.2.1.细胞株HEK293T的复苏与培养
取液氮冻存的HEK293T细胞,放于37℃水浴中解冻;解冻后800rpm条件下离心2min迅速加入1ml体积分数为10%胎牛血清的DMEM培养液中(DMEM培养液需要提前预热至37℃),枪头轻轻吹打至无细胞团存在;再将离心管中细胞悬液转移至8ml提前预热至37℃体积分数为10%胎牛血清的DMEM培养液的10cm培养皿中,均匀分布细胞后,并于37℃、含5%的CO2培养箱中培养24h,以后每隔2天换液至细胞密度达90%时传代。
4.2.2.质粒DNA转染及慢病毒收集
转染前一天3x106 HEK293T细胞接种到10cm平皿中,培养12h后细胞约有70%融合,转染前4h,将含血清的培养液换为无双抗无血清的opti-MEM(5ml/孔);Lipofectamine2000(Invitrogen)作为转染试剂将10ug lentiCRISPRv2-sgRNA/7ug psPAX2/3ug VSV-G转染至HEK293T细胞,具体步骤依照说明书进行(所有试剂均为一个孔的试剂用量):用opti-MEM培养液将20ug无内毒素的质粒稀释至500ul,室温孵育;同样用opti-MEM培养液将20ul Lipofectamine2000稀释至500ul;将孵育的质粒和Lipofectamine2000混匀,总体积为1000ul,混合溶液室温孵育30min;将1000ul混合液加到培养板的一个孔中,轻轻晃动培养板混匀培养液;将细胞置于37℃、含5%的C02培养箱中培养,6h后将培养液换为体积分数为10%胎牛血清的DMEM培养液继续培养18h,更换10ml新鲜培养基。24h后收取细胞上清并更换新鲜培养基,48h后再次收集上清。
4.2.3.慢病毒纯化
病毒上清用40um滤器过滤去除细胞碎片;将过滤后的上清转移至超滤柱(Merck Millipore)上;4℃,4000rpm离心30分钟,去除滤液获得纯化的病毒。
4.2.4.慢病毒滴度测定
荧光定量PCR测定慢病毒滴度,按照qPCR Lentivirus Titration试剂盒(abm)说明书操作。
4.3.慢病毒感染L929细胞
4.3.1.细胞株L929的复苏与培养
取液氮冻存的L929细胞,放于37℃水浴中解冻;解冻后800rpm条件下离心2min迅速加入1ml体积分数为10%胎牛血清的DMEM培养液中(DMEM培养液需要提前预热至37℃),枪头轻轻吹打至无细胞团存在;再将离心管中细胞悬液转移至8ml提前预热至37℃体积分数为10%胎牛血清的DMEM培养液的10cm培养皿中,均匀分布细胞后,并于37℃、含5%的CO2培养箱中培养24h,以后每隔2天换液至细胞密度达90%时传代。
4.3.2.慢病毒感染L929
按照每孔1x105细胞接种到12孔板,培养12h后细胞约有50%融合,将原有培养液换为含有慢病毒的新鲜培养基;将细胞置于37℃、含5%的CO2培养箱中培养24h至36h。
4.3.3.慢病毒感染阳性L929细胞筛选
感染结束后的细胞传代于6孔板,培养3h后至细胞完全贴壁;将原有的培养基更换为含有5ug/ml嘌呤霉素培养基(体积分数为10%胎牛血清的DMEM培养液)中培养至融合度95%后传代培养。连续传代4次。
4.4.小鼠RIP1敲除效率的检测
4.4.1.蛋白印迹实验检测小鼠RIP1蛋白水平
用RIPA裂解液裂解步骤4.3获得的细胞并定量,取20ug蛋白样品上样进行SDS-PAGE电泳,转膜后与anti-RIP1(BD)4℃孵育过夜后与抗鼠二抗室温孵育1h后显影曝光,结果如图4所示,图中,N表示原始细胞,不经过任何感染的细胞;Lo exp短时间曝光,条带浅;Hi exp长时间曝光,条带深。
4.4.2.诱导细胞坏死条件下鉴定小鼠RIP1是否敲除
按照每孔1x104细胞接种到96孔板,培养3h后细胞贴壁,去除培养液后分别加入含二甲基亚砜(DMSO)或TNFα+zVAD的培养液。将细胞置于37℃、含5%的CO2培养箱中培养,24h后利用细胞存活检测试剂盒(Cell Titer-Glo Luminescent Cell Viability Assay kit,Promega)检测细胞存活情况,结果如图5 所示。图中,N表示原始细胞,不经过任何感染的细胞。
虽然以具体实施例的方式阐述了本发明,但应理解,本发明并不限于上述内容。在不偏离本发明精神的情况下对本发明作出的各种修改和变动,均在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!